![]()
scadnano ("scriptable-cadnano") is a program for designing synthetic DNA structures such as DNA origami. The scadnano Python package (source code repository here) is a library for programmatically creating and editing these nanostructures. The scadnano project is developed and maintained by the UC Davis Molecular Computing group. Note that cadnano is a separate project, developed and maintained by the Douglas lab at UCSF.
If you find scadnano useful in a scientific project, please cite its associated paper:
scadnano: A browser-based, scriptable tool for designing DNA nanostructures.
David Doty, Benjamin L Lee, and Tristan Stérin.
DNA 2020: Proceedings of the 26th International Conference on DNA Computing and Molecular Programming
[ paper | BibTeX ]
Note: If you are reading this on the PyPI website, some of the links below won't work. Many are relative links intended to be read on the GitHub README page.
- Overview
- Reporting issues
- Installation
- Example
- Abbreviated syntax with chained methods
- StrandBuilder object for iteratively building up strands with many domains
- Tutorial
- API documentation
- Other examples
- Contributing
This package is used to write Python scripts outputting .sc files readable by scadnano, a web application useful for displaying and manually editing these structures. The purpose of this module is to help automate some of the task of creating DNA designs, as well as making large-scale changes to them that are easier to describe programmatically than to do by hand in the scadnano web interface.
We will try to announce breaking changes (and possibly new features) under the GitHub releases page. The version numbers in this Python library repo and the web interface repo won't always advance at the same time, and sometimes a feature is supported in one before the other.
Following semantic versioning, version numbers are major.minor.patch, i.e., version 0.9.2 has minor version number 9. Prior to version 1.0.0, when a breaking change is made, this will increment the minor version (for example, going from 0.9.4 to 0.10.0). After version 1.0.0, breaking changes will increment the major version.
Please report issues in the web interface at the scadnano web interface GitHub repository, and report issues in the Python scripting library at the scadnano Python package GitHub repository.
Short version: type this at the command line:
pip install scadnanoRead below for troubleshooting suggestions if that didn't work.
The scadnano Python package requires Python version 3.7 or later.
To check your current version of Python, open a command line and type
python --version
If it is version 2.7 or below, type
python3 --version
If that fails, or reports Python version 3.6 or below, you will have to install a later version of Python. Follow this link to install Python. You may also use an alternative Python distribution, such as Anaconda.
Once Python is installed, there are two ways you can install the scadnano Python package:
-
pip (recommended)
Use pip to install the package by executing the following at the command line:
pip install scadnanoIf it worked, you should be able to open a Python interpreter and import the scadnano module:
Python 3.7.9 (default, Aug 31 2020, 17:10:11) [MSC v.1916 64 bit (AMD64)] :: Anaconda, Inc. on win32 Type "help", "copyright", "credits" or "license" for more information. >>> import scadnano as sc >>> print(sc.Domain(helix=1, forward=True, start=0, end=8)) Domain(, helix=1, forward=True, start=0, end=8) >>>
If the above does not work for you, here are some things to try.
If your Python installation does not already have pip installed, you may have to install it. Executing this Python script should work; see also https://docs.python.org/3/installing/index.html or https://www.liquidweb.com/kb/install-pip-windows/.
Once pip is installed, or if you believe it is already installed, check your version of
pipby typingpip --versionIt should say something like
pip 19.3.1 from ...lib\site-packages\pip (python 3.8)If the version of Python at the end is Python 3.7 or higher, you are good. If it is version 2.7 or lower, type
pip3 --versionIf that works and shows Python 3.7 or higher, you are good, but you should type
pip3in the subsequent instructions instead ofpip.If it shows Python 3.6, install the dataclasses backport module via
pip install dataclassesIf it shows Python 3.5 or lower, then you will need to upgrade your Python version (recommended Python 3.7 or higher).
-
download
As a simple alternative (in case you run into trouble using pip), you can simply download the scadnano.py file. However, you need to first install two packages that are required by scadnano: Install openpyxl and tabulate by typing the following at the command line:
pip install openpyxl tabulate.Download and place the following files in your PYTHONPATH (e.g., in the same directory as the scripts you are running). Note: If you are reading this on the PyPI website or anywhere other than GitHub, the links below won't work. They are relative links intended to be read on the GitHub README page.
- required: scadnano.py
- optional: modifications.py; This contains some common DNA modifications such as biotin and Cy3.
- optional: origami_rectangle.py; This can help create origami rectangles, but it is not necessary to use scadnano.
To download them, right-click on "Raw" near the top and select (in Chrome or Firefox) "Save link as...":

The scadnano package uses the Python package xlwt to write Excel files, so xlwt must be installed in order to call the method
Design.write_idt_plate_excel_file()to export an Excel file with DNA sequences. To install xlwt, typepip install xlwtat the command line. (If you instead use pip to install the scadnano package, xlwt will be automatically installed.)

Consider the following design:
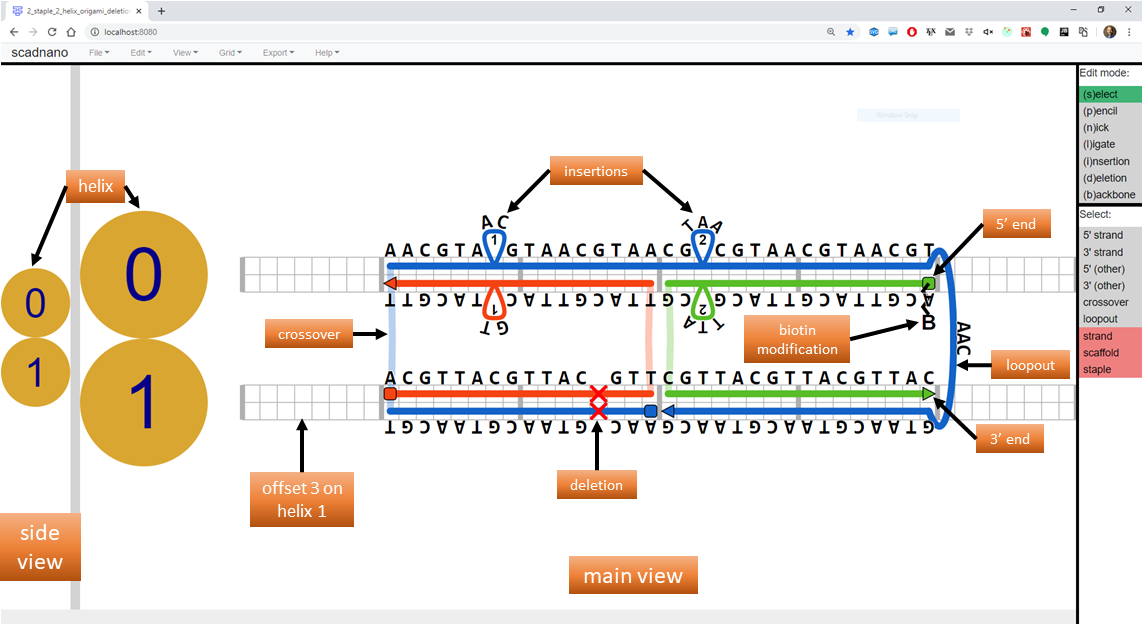
The following Python script produces this design.
import scadnano as sc
import modifications as mod
def create_design() -> sc.Design:
# helices
helices = [sc.Helix(max_offset=48), sc.Helix(max_offset=48)]
# left staple
stap_left_domain1 = sc.Domain(helix=1, forward=True, start=8, end=24)
stap_left_domain0 = sc.Domain(helix=0, forward=False, start=8, end=24)
stap_left = sc.Strand(domains=[stap_left_domain1, stap_left_domain0])
# right staple
stap_right_domain0 = sc.Domain(helix=0, forward=False, start=24, end=40)
stap_right_domain1 = sc.Domain(helix=1, forward=True, start=24, end=40)
stap_right = sc.Strand(domains=[stap_right_domain0, stap_right_domain1])
stap_right.set_modification_5p(mod.biotin_5p)
# scaffold
scaf_domain1_left = sc.Domain(helix=1, forward=False, start=8, end=24)
scaf_domain0 = sc.Domain(helix=0, forward=True, start=8, end=40)
loopout = sc.Loopout(length=3)
scaf_domain1_right = sc.Domain(helix=1, forward=False, start=24, end=40)
scaf = sc.Strand(domains=[scaf_domain1_left, scaf_domain0, loopout, scaf_domain1_right], is_scaffold=True)
# whole design
design = sc.Design(helices=helices, strands=[scaf, stap_left, stap_right], grid=sc.square)
# deletions and insertions added to design are added to both strands on a helix
design.add_deletion(helix=1, offset=20)
design.add_insertion(helix=0, offset=14, length=1)
design.add_insertion(helix=0, offset=26, length=2)
# also assigns complement to strands other than scaf bound to it
design.assign_dna(scaf, 'AACGT' * 18)
return design
if __name__ == '__main__':
design = create_design()
design.write_scadnano_file(directory='output_designs')Running the code above produces a .sc file that, if loaded into scadnano, should appear as in the screenshot above. The web interface README explains many of the terms used in the code (domain, helix, loopout, insertion, etc.).
Instead of explicitly creating variables and objects representing each domain in each strand, there is a shorter syntax using chained method calls. Instead of the above, create only the helices first, then create the Design. Then strands can be added using a shorter syntax, to describe how to draw the strand starting at the 5' end and moving to the 3' end. The following is a modified version of the above create_design function using these chained methods:
def create_design() -> sc.Design:
# helices
helices = [sc.Helix(max_offset=48), sc.Helix(max_offset=48)]
# whole design
design = sc.Design(helices=helices, grid=sc.square)
# for absolute offsets, call method "to"
# left staple
design.draw_strand(1, 8).to(24).cross(0).to(8)
# for relative offsets, call method "move"
# right staple
design.draw_strand(0, 40).move(-16).cross(1).move(16).with_modification_5p(mod.biotin_5p)
# scaffold
design.draw_strand(1, 24).move(-16).cross(0).move(32).loopout(1, 3).move(-16).as_scaffold()
# deletions and insertions added to design are added to both strands on a helix
design.add_deletion(helix=1, offset=20)
design.add_insertion(helix=0, offset=14, length=1)
design.add_insertion(helix=0, offset=26, length=2)
# also assigns complement to strands other than scaf bound to it
design.assign_dna(design.scaffold, 'AACGT' * 18)
return designDocumentation is available in the API docs.
The method Design.draw_strand, as well as all those that follow it in a chained method call (e.g., move, cross, etc.) all return an instance of the class StrandBuilder.
Above, that StrandBuilder instance is anonymous, i.e., never assigned to a variable.
Some long strands may be easier to specify with loops, for example an M13 scaffold strand for an origami.
If so, then to use the above methods, assign the StrandBuilder object to a variable, and call the relevant methods on that object to build up the strand in each iteration of the loop.
For example, the following modification of the above create_design function creates a linear scaffold strand that zig-zags back and forth across 32 helices:
def create_design() -> sc.Design:
num_helices = 32
helices = [sc.Helix(max_offset=200) for _ in range(num_helices)]
design = sc.Design(helices=helices, grid=sc.square)
strand_builder = design.draw_strand(0, 0)
for helix in range(num_helices):
# move forward if on an even helix, otherwise move in reverse
move_distance = 200 if helix % 2 == 0 else -200
strand_builder.move(move_distance)
if helix < 31: # crossover to next helix, unless it's the last helix
strand_builder.cross(helix + 1)
strand_builder.as_scaffold()
return designOnline documentation of the package API (which classes, methods, functions, and constants are provided by the package) is located here: https://scadnano-python-package.readthedocs.io
A tutorial shows how to create a "standard" 24-helix DNA origami rectangle using the scadnano Python package.
Note: If you are reading this on the PyPI website, the links below won't work. They are relative links intended to be read on the GitHub README page.
Several example scripts are located in the examples/ subfolder. Their output is contained in the examples/output_designs/ subfolder.
If you wish to contribute to scadnano, please see the CONTRIBUTING document to contribute to the scadnano Python package. There is also a CONTRIBUTING document for the web interface.