Serpentine is a project that leverages the snakemake workflow management to create a flexible, efficient, and highly parallelizable pipelines for data analysis. The current focus is on DNA sequence analysis, but there is no limitation on the workflows that can be implemented. We have definite plans for RNA-seq and combined RNA/DNA pipelines in the works. Additional pipeline suggestions are welcome.
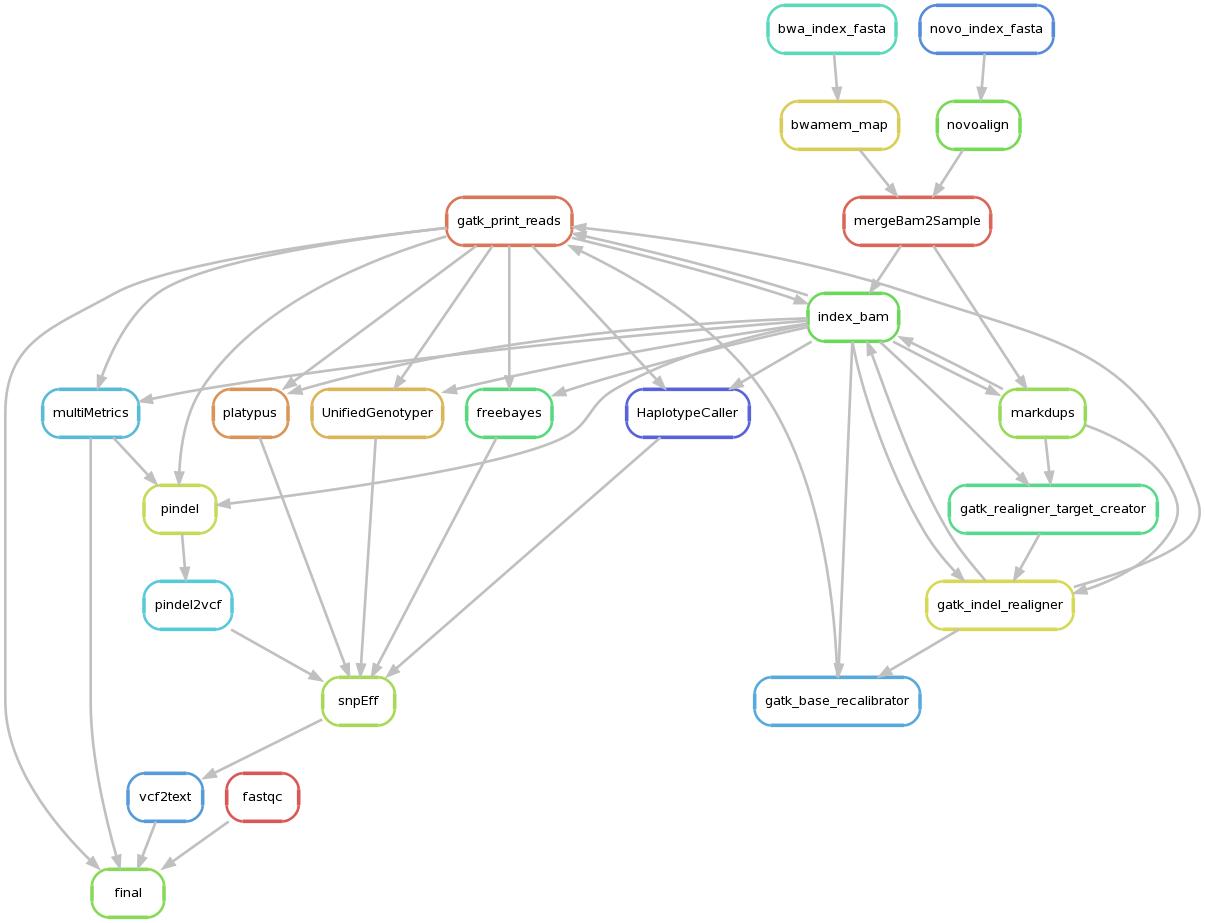
Serpentine can currently run workflows composed of any combination of the following steps. The steps are run in a fully-parallel manner based on job dependencies and available resources. The whole process is driven by a json config file (example config file).
- aligners:
- novoalign
- bwa-mem
- postprocessing:
- Duplicate marking
- realignment around indels
- base quality recalibration
- variant callers:
- germline:
- UnifiedGenotyper
- HaplotypeCaller
- Freebayes
- platypus
- pindel
- germline:
- metrics:
- insert size
- quality metrics
- alignment summary metrics
- duplication metrics
=======
The easiest way to get serpentine is to clone the repository.
git clone https://github.com/NCI-CCR-GB/serpentine.git
To contribute to the project, you can:
If you have questions, file a new issue and label as a question or email me.
- Sample names cannot have "/" or "." in them
- fastq files end in ".fastq.gz"
An example config file is available here.