Home
Welcome to the wiki for the tool for Distilling and Refining Annotations of Metabolism (DRAM)! Here you will find all you need to know to setup, install and run DRAM and DRAM-v. This page will give you basic instructions to rapidly get DRAM installed and working. If you want more detailed instructions on how DRAM works or what all DRAM options mean then be sure to check out the other pages in the DRAM wiki (see sidebar).
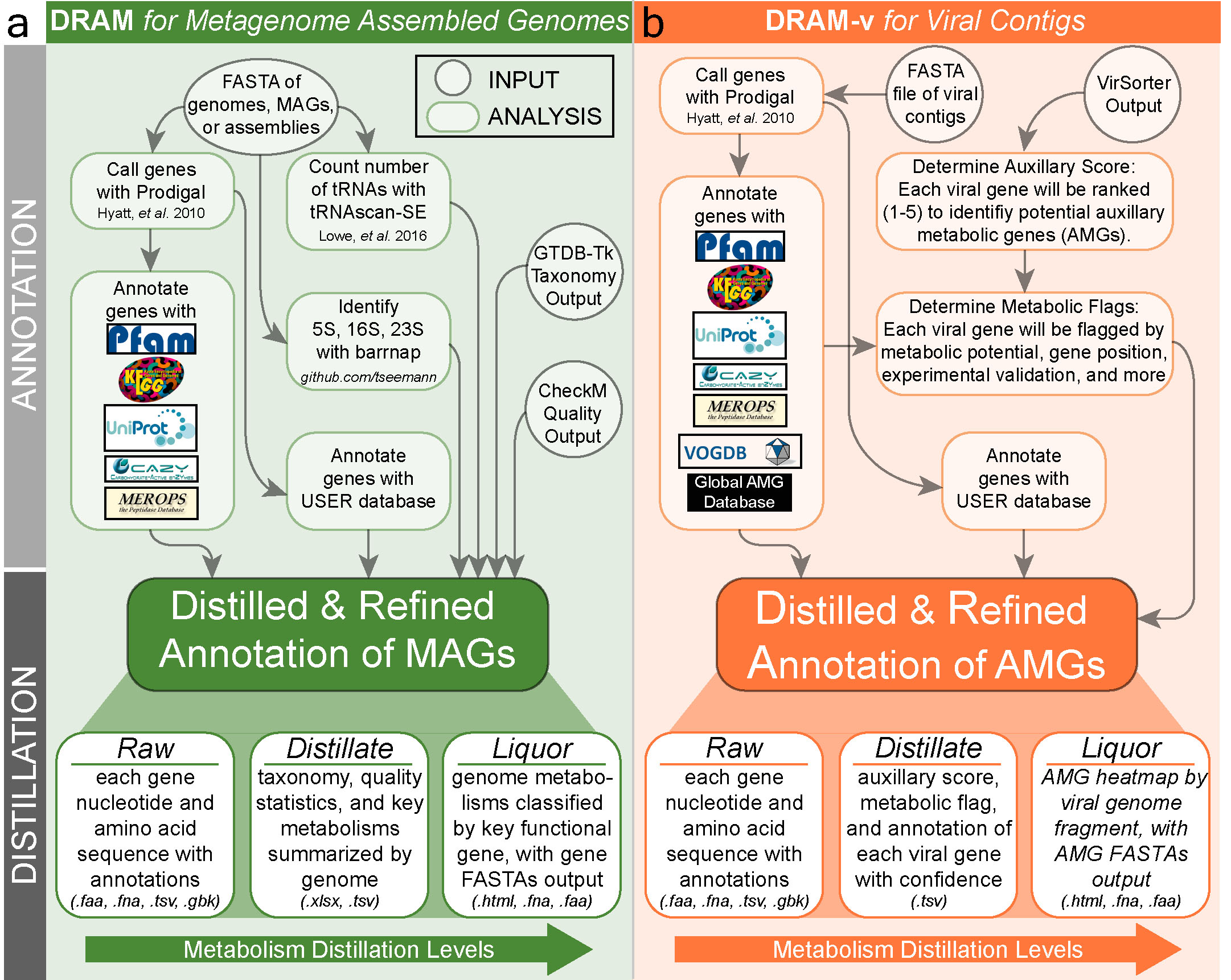
Like the process of making the eponymous glass of whiskey, DRAM distills genome annotations to metabolic functions in three levels that scale in information: (1) Raw, (2) Distillate, and (3) Liquor. Through this distillation process, DRAM is able to annotate high volumes of microbial genomes and organize the resulting information in a way that highlights functional guilds, allowing users to infer organismal metabolism across hundreds of genomes. To obtain the Raw output, DRAM calls genes on input genomes, searches each gene against seven databases, and considers all derived annotations together. This approach significantly increases database searches by at least 25% beyond other annotators such as DFAST, MetaERG, and Prokka. The DRAM Raw output contains all database hits per gene in every input genome, which is the final output for most annotators. DRAM significantly advances genome annotation beyond the final raw output by providing the first of its kind organization and visualization of all annotations into ecosystem relevant functions.
DRAM for MAGs works by annotating all genomes given with all databases used by DRAM. The user is given a tab delimited annotations file with all annotations from all databases for all genes in all genomes. Additionally the user is given a folder with genbank files for each genome, a gff file with all annotations across genomes as well as annotated nucleotide and amino acid fasta files of all genes. The results of annotation can be distilled. This will generate three files: 1. The genome statistics table which includes all statistics required by MIMAG, 2. the metabolism summary which gives gene counts of functional genes across a wide variety of metabolisms and 3. the liquor which is a heatmap showing coverage of modules, the coverage of electron transport chain components and the presence of selected metabolic functions.
DRAM-v works to annotate and discover potential auxiliary metabolic genes (AMGs) from viral contigs as detected by VirSorter. The output of VirSorter is first used for annotation using the same databases as in DRAM for MAGs. After that auxiliary scores, which measure the confidence that a gene is viral in origin, and metabolic flags, which indicate different characteristics of each gene, are generated. Based on the auxiliary scores and metabolic flags potential AMGs are identified. By default a gene is considered a potential AMG if it has an M flag, no V flag, no A flag and an auxiliary score of 3 or lower. Distillation of viral annotations generates: 1. A viral genome statistics table which includes all statistics required by MIUViG, 2. a summary of the potential AMGs present in all viral contigs as well as their metabolic functions and 3. the liquor which is a heatmap showing the functions of all AMGs present across all viral contigs.
These are the commands needed to quickly install, setup and get started running DRAM and DRAM-v.
It is recommended to install DRAM within a conda environment. If you would like to install DRAM manually see the How to Install and Set Up DRAM section of the Wiki.
wget https://raw.githubusercontent.com/WrightonLabCSU/DRAM/master/environment.yaml
conda env create -f environment.yaml -n DRAM
git clone https://github.com/WrightonLabCSU/DRAM.git
pip install -e .If this installation method is used then all further steps should be run inside the created DRAM environment.
Then set up DRAM using the following command:
DRAM-setup.py prepare_databases --output_dir DRAM_data --kegg_loc kegg.pepOnce DRAM is set up you are ready to annotate some MAGs. The following commands will generate the full annotation and distillation of genomes:
DRAM.py annotate -i 'my_bins/*.fa' -o annotation
DRAM.py distill -i annotation/annotations.tsv -o distill --trna_path annotation/trnas.tsv --rrna_path annotation/rrnas.tsvAnnotating and distilling viral contigs requires some preprocessing and an additional input. The contigs must be processed with VirSorter and the processed viral contigs and VIRSorter_affi-contigs.tab are used as input to DRAM-v. The following commands will generate the full annotation and distillation of viral contigs:
DRAM-v.py annotate -i my_viral_contigs.fa -v VIRSorter_affi-contigs.tab -o annotation
DRAM-v.py distill -i annotation/annotations.tsv -o annotation/distilledDRAM has a large memory burden and is designed to be run on high performance computers. DRAM annotates against a large variety of databases which must be processed and stored. Setting up DRAM with KEGG Genes and UniRef90 will take up ~500 GB of storage after processing and require ~512 GB of RAM while using KOfam and skipping UniRef90 will mean all processed databases will take up ~30 GB on disk and will only use ~128 GB of RAM while processing. DRAM annotation memory usage depends on the databases used. When annotating with UniRef90 around 220 GB of RAM is required. If the KEGG gene database has been provided and UniRef90 is not used then memory usage is around 100 GB of RAM. If KOfam is used to annotate KEGG and UniRef90 is not used then less than 50 GB of RAM is required. DRAM can be run with any number of processors on a single node.
DRAM annotate functions are only tested in linux and DRAM distillation functions are tested in linux and macOS environments. It will likely work in OSX environments. It is unlikely to work in Windows.