



![]()
![]()
![]()
![]()
![]()
This repository contains python toolset for Structure-Informed Property and Feature Engineering with Neural Networks which implements a numer of user-friendly tools for:
- Calculating different vector representations of atomic structures for a number of applications including supervised (e.g., predictive machine learning models) and unsupervised learning (e.g., clustering of atomic structures based on similarity or performing anomaly detection). Notably, utilize crystallographic information and some other techniques to make this process very efficient for the vast majority of use cases (see arXiv:2404.02849)
- Efficient deployment of pre-trained ML models (not limited to neural networks) obtained from repositories like Zenodo (including some we trained) or trained locally on user's machine. The system is very plug-and-play thanks to using Open Neural Network Exchange (ONNX) format which can be exported from nearly any machine learning framework.
- Tuning pre-trained ML models to new domains, like new chemical compositions, different ab initio functional, or entirely new properties. Since V0.16, users can take advantage of integration with OPTIMADE API which allows one to tune models based on DFT datasets like Materials Project, OQMD, AFLOW, or NIST-JARVIS, in just 3 lines of code specifying which provider to use, what to query for, and hyperparameters for tuning.
The underlying methodology, efficiency optimizations, design choices, and implementation specifics are given in the following publications:
-
Adam M. Krajewski, Jonathan W. Siegel, Zi-Kui Liu, Efficient Structure-Informed Featurization and Property Prediction of Ordered, Dilute, and Random Atomic Structures, April 2024, arXiv:2404.02849
-
Adam M. Krajewski, Jonathan W. Siegel, Jinchao Xu, Zi-Kui Liu, Extensible Structure-Informed Prediction of Formation Energy with improved accuracy and usability employing neural networks, Computational Materials Science, Volume 208, 2022, 111254, DOI:10.1016/j.commatsci.2022.111254
A more complete (and verbose) description of capabilities is given in documentation at (pysipfenn.org). You may also consider visiting our Phases Research Lab website at (phaseslab.org).
-
(v0.16.0) Three exciting news! (1) The all new
ModelAdjusterssubmodule automates tuning and can fetch data directly fromOPTIMADE API; (2) A new manuscript detailing advantages of our featurization tools has been put on arXiv:2404.02849; and (3) the name of the software was updated to python toolset for Structure-Informed Property and Feature Engineering with Neural Networks to retain thepySIPFENNacronym but better reflect our strengths and development direction. -
(v0.15.0) A new descriptor (feature vector) calculator
KS2022_randomSolutionshas been implemented. It is used for structure-informed featurization of compositions randomly occupying a lattice, spiritually similar to SQS generation, but also taking into account (1) chemical differences between elements and (2) structural effects. -
(v0.14.0) Users can now take advantage of a Prototype Library to obtain common structures from any
Calculatorinstance withc.prototypeLibrary[<name>]['structure']. It can be easily updated or appended with high-level API or by manually modifyig its YAML here. -
(v0.13.0) Model exports (and more!) to PyTorch, CoreML, and ONNX are now effortless thanks to
core.modelExportersmodule. Please note you need to install pySIPFENN withdevoption (e.g.,pip install "pysipfenn[dev]") to use it. See docs here. -
(v0.12.2) Swith to LGPLv3 allowing for integration with proprietary software developed by CALPHAD community, while supporting the development of new pySIPFENN features for all.
-
(March 2023 Workshop) We would like to thank all 100 of our amazing attendees for making our workshop, co-organized with the Materials Genome Foundation.
The figure below is the main schematic of pySIPFENN framework detailing the interplay of internal components. The user interface provides a high-level API to process structural data within core.Calculator, pass it to featurization submodules in descriptorDefinitions to obtain vector representation, then passed to models defined in models.json and (typically) run automatically through all available models. All internal data of core.Calculator is accessible directly, enabling rapid customization. An auxiliary high-level API enables advanced users to operate and retrain the models.
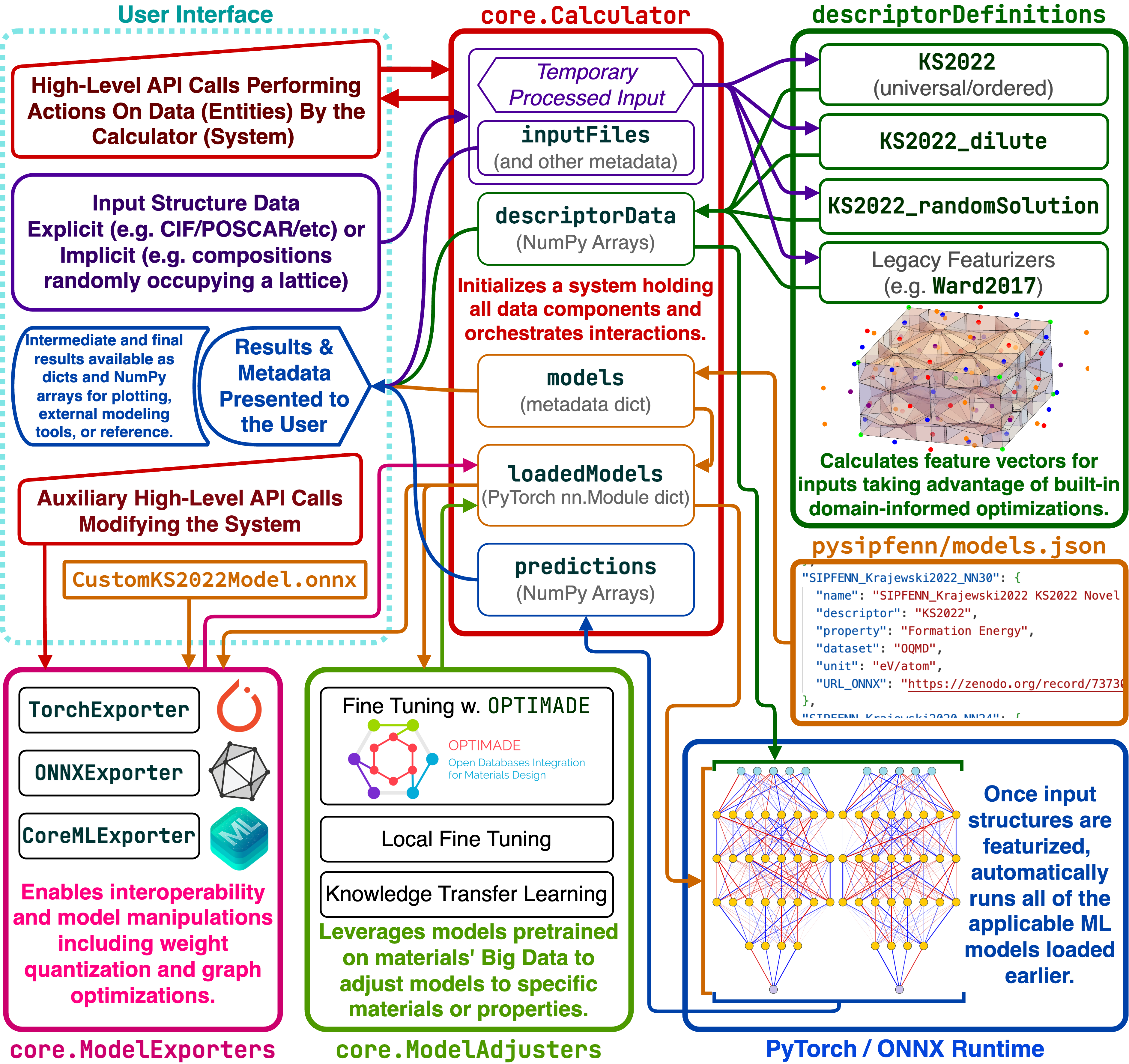
pySIPFENN is a very flexible tool that can, in principle, be used for the prediction of any property of interest that depends on an atomic configuration with very few modifications. The models shipped by default are trained to predict formation energy because that is what our research group is interested in; however, if one wanted to predict Poisson’s ratio and trained a model based on the same features, adding it would take minutes. Simply add the model in open ONNX format and link it using the models.json file, as described in the documentation.
In our line of work, pySIPFENN and the formation energies it predicts are usually used as a computational engine that generates proto-data for creation of thermodynamic databases (TDBs) using ESPEI (https://espei.org). The TDBs are then used through pycalphad (https://pycalphad.org) to predict phase diagrams and other thermodynamic properties.
Another of its uses in our research is guiding the Density Functional Theory (DFT) calculations as a low-cost screening tool. Their efficient conjunction then drives the experiments leading to discovery of new materials, as presented in these two papers:
-
Sanghyeok Im, Shun-Li Shang, Nathan D. Smith, Adam M. Krajewski, Timothy Lichtenstein, Hui Sun, Brandon J. Bocklund, Zi-Kui Liu, Hojong Kim, Thermodynamic properties of the Nd-Bi system via emf measurements, DFT calculations, machine learning, and CALPHAD modeling, Acta Materialia, Volume 223, 2022, 117448, https://doi.org/10.1016/j.actamat.2021.117448.
-
Shun-Li Shang, Hui Sun, Bo Pan, Yi Wang, Adam M. Krajewski, Mihaela Banu, Jingjing Li & Zi-Kui Liu, Forming mechanism of equilibrium and non-equilibrium metallurgical phases in dissimilar aluminum/steel (Al–Fe) joints. Nature Scientific Reports 11, 24251 (2021). https://doi.org/10.1038/s41598-021-03578-0
Installing pySIPFENN is simple and easy by utilizing PyPI package repository, conda-forge package repository, or by cloning from GitHub directly.
While not required, it is recommended to first set up a virtual environment using venv or Conda. This ensures that (a) one of the required
versions of Python (3.9+) is used and (b) there are no dependency conflicts. If you have Conda installed on your system (see miniconda install instructions), you can create a new environment with a simple:
conda create -n pysipfenn python=3.10 jupyter numpy
conda activate pysipfenn
If you are managing a large set of dependencies in your project, you may consider using mamba in place of conda. It is a less mature, but much faster drop-in replacement compatible with existing environments. See micromamba install instructions.
If your main goal is to run pySIPFENN models, provided by us or any other vendor, you need only a subset of the capabilities of our code, so you can follow with the following install. Simply install pySIPFENN:
-
from PyPI with
pip:pip install pysipfenn
-
from conda-forge with
conda:conda install -c conda-forge pysipfenn
-
from conda-forge with
micromamba:micromamba install -c conda-forge pysipfenn
-
from source, by cloning. To get a stable version, you can specify a version tag after the URL with
--branch <tag_name> --single-branch, or omit it to get the development version (which may have bugs!):git clone https://github.com/PhasesResearchLab/pySIPFENN.git
then move to
pySIPFENNdirectory and install in editable (-e) mode.cd pySIPFENN pip install -e .
If you want to utilize pySIPFENN beyond its core functionalities, for instance, to train new models on custom datasets or to export models in different
formats or precisions, you need to install several other dependencies. This can be done by following the from source instructions above but appending
the last instruction with dev extras marker.
pip install -e ".[dev]"Note:
pip install "pysipfenn[dev]"will also work, but will be less conveninet for model modifications (which you likely want to do), as all persisted files will be located outside your working directory. You can quickly find where, by callingimport pysipfenn; c = pysipfenn.Calculator(); print(c)andCalculatorwill tell you (amongst other things) where they are.
If you wish to contribute to the development of pySIPFENN you are more than welcome to do so by forking the repository and creating a pull request. As of Spring 2024, we are actively developing the code and we should get back to you within a few days. We are also open to collaborations and partnerships, so if you have an idea for a new feature or a new model, please do not hesitate to contact us through the GitHub issues or by email.
In particular, we are seeking contributions in the following areas:
-
New Models: We are always looking for new models to add to the repository. We have several (yet) unpublished ones for several different properties, so there is a good chance it will work for your case as well. We are happy to provide basic support for training, including using the default model for transfer learning on small datasets.
-
New Featurizers / Descriptor Sets: We are always looking for new ways to featurize atomic configurations.
- We are particularly interested in including more domain-specific knowledge for different niches of materials science. Our KS2022 does a good job for most materials, but we look to expand it.
- We are not looking for featurizers that (a) cannot embed a structure into the feature space (e.g., most of the graph representations, which became popular in the last two years) or (b) do not encode physics into the feature space (e.g., raw atomic coordinates or 3D voxel representations).
- Note: Autoencoders which utilize graph or 3D voxel representations to encode latent space position to predict property/properties fall into the first category and are very welcome.
-
Quality of Life Improvements: We are always looking for ways to make the software easier to use and more efficient for users. If you have an idea for a new data parsing method, or a new way to visualize the results, we would love to hear about it.
We are currently very flexible with the rules for contributing, despite being quite opinionated :)
Some general guidelines are:
-
The
coremodule is the only one that should be used by our typical end user. All top-level APIs should be defined in thepysipfenn.pythrough theCalculatorclass. APIs operating on theCalculatorclass, to export or retrain models, should be defined outside it, but withinpysipfenn.coremodule. -
All featurizers / descriptor calculators must be self-contained in a single submodule (file or directory) of
pysipfenn.descriptorDefinitions(i.e., not spread around the codebase) and depend only on standard Python library and current pySIPFENN dependencies, includingnumpy,torch,pymatgen,onnx,tqdm. If you need to add a new dependency, please discuss it with us first. -
All models must be ONNX models, which can be obtained from almost any machine learning framework. We are happy to help with this process.
-
All new classes, attributes, and methods must be type-annotated. We are happy to help with this process.
-
All new classes, attributes, and methods must have a well-styled docstring. We are happy to help with this process.
-
All functions, classes, and methods should have explicit inputs, rather than passing a dictionary of parameters (*kwargs). This does require a bit more typing, but it makes the code much easier to use for the end user, who can see in the IDE exactly what parameters are available and what they do.
-
All functions, classes, and methods should explain why they are doing something, not just what they are doing. This is critical for end-users who did not write the code and are trying to understand it. In particular, the default values of parameters should be explained in the docstring.
-
All new features must be tested with the
pytestframework. Coverage should be 100% for new code or close to it for good reasons. We are happy to help with this process.
If you use pySIPFENN software, please consider citing:
-
Adam M. Krajewski, Jonathan W. Siegel, Zi-Kui Liu, Efficient Structure-Informed Featurization and Property Prediction of Ordered, Dilute, and Random Atomic Structures, April 2024, arXiv:2404.02849
-
Adam M. Krajewski, Jonathan W. Siegel, Jinchao Xu, Zi-Kui Liu, Extensible Structure-Informed Prediction of Formation Energy with improved accuracy and usability employing neural networks, Computational Materials Science, Volume 208, 2022, 111254, DOI:10.1016/j.commatsci.2022.111254
If you are using predictions from pySIPFENN models accessed through OPTIMADE from MPDD, please additionally cite:
- Matthew L. Evans, Johan Bergsma, ..., Adam M. Krajewski, ..., Zi-Kui Liu, ..., et al., Developments and applications of the OPTIMADE API for materials discovery, design, and data exchange, 2024, arXiv:2402.00572