diff --git a/.github/workflows/integration-test.yml b/.github/workflows/integration-test.yml
index e2805c72..3bdc8150 100644
--- a/.github/workflows/integration-test.yml
+++ b/.github/workflows/integration-test.yml
@@ -56,6 +56,28 @@ jobs:
uses: pre-commit/action@v3.0.1
with:
extra_args: --all-files
+ python-type-checks:
+ # This job is used to check Python types
+ name: Python type checks
+ # Avoid fail-fast to retain output
+ strategy:
+ fail-fast: false
+ runs-on: ubuntu-22.04
+ if: github.event_name != 'schedule'
+ steps:
+ - name: Checkout repo
+ uses: actions/checkout@v4
+ - name: Setup python, and check pre-commit cache
+ uses: ./.github/actions/setup-env
+ with:
+ python-version: ${{ env.TARGET_PYTHON_VERSION }}
+ cache-pre-commit: false
+ cache-venv: true
+ setup-poetry: true
+ install-deps: true
+ - name: Run mypy
+ run: |
+ poetry run mypy .
integration-test:
name: Pytest (Python ${{ matrix.python-version }} on ${{ matrix.os }})
# Runs pytest on all tested versions of python and OSes
diff --git a/README.md b/README.md
index c7dd62f5..c621c4c0 100644
--- a/README.md
+++ b/README.md
@@ -11,16 +11,20 @@
Pycytominer is a suite of common functions used to process high dimensional readouts from high-throughput cell experiments.
The tool is most often used for processing data through the following pipeline:
- +
+
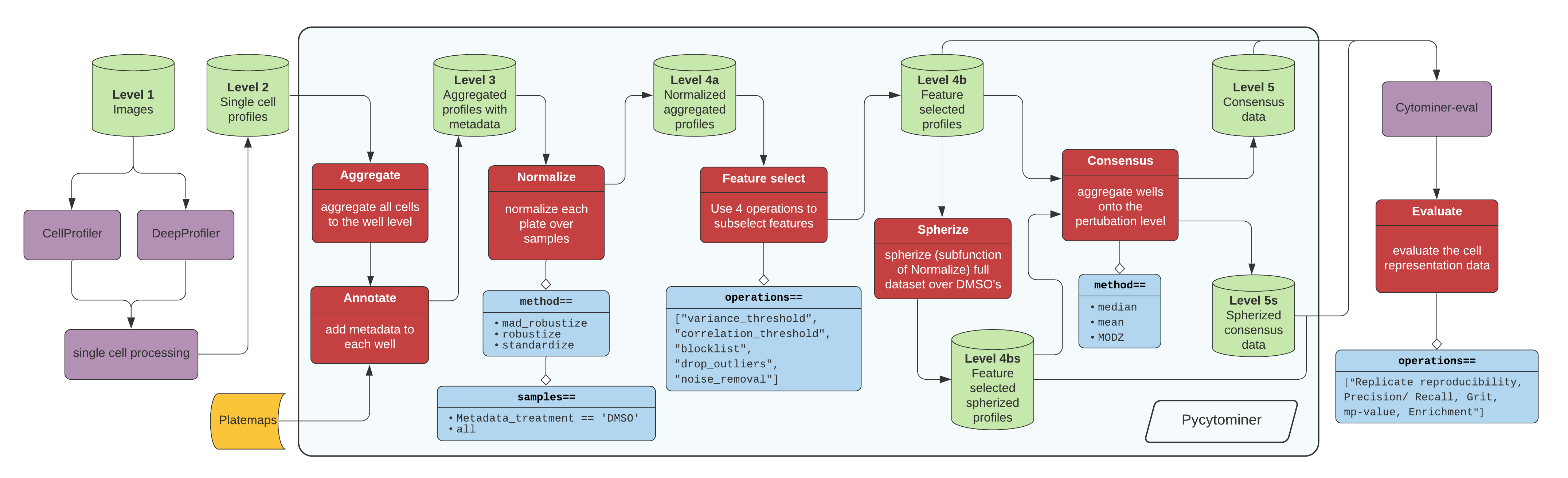
+> Figure 1. The standard image-based profiling experiment and the role of Pycytominer. (A) In the experimental phase, a scientist plates cells, often perturbing them with chemical or genetic agents and performs microscopy imaging. In image analysis, using CellProfiler for example, a scientist applies several data processing steps to generate image-based profiles. In addition, scientists can apply a more flexible approach by using deep learning models, such as DeepProfiler, to generate image-based profiles. (B) Pycytominer performs image-based profiling to process morphology features and make them ready for downstream analyses. (C) Pycytominer performs five fundamental functions, each implemented with a simple and intuitive API. Each function enables a user to implement various methods for executing operations.
[Click here for high resolution pipeline image](https://github.com/cytomining/pycytominer/blob/main/media/pipeline.png)
-Image data flow from a microscope to cell segmentation and feature extraction tools (e.g. CellProfiler or DeepProfiler).
+Image data flow from a microscope to cell segmentation and feature extraction tools (e.g. [CellProfiler](https://cellprofiler.org/) or [DeepProfiler](https://cytomining.github.io/DeepProfiler-handbook/docs/00-welcome.html)) (**Figure 1A**).
From here, additional single cell processing tools curate the single cell readouts into a form manageable for pycytominer input.
-For CellProfiler, we use [cytominer-database](https://github.com/cytomining/cytominer-database) or [CytoTable](https://github.com/cytomining/CytoTable).
-For DeepProfiler, we include single cell processing tools in [pycytominer.cyto_utils](pycytominer/cyto_utils/).
+For [CellProfiler](https://cellprofiler.org/), we use [cytominer-database](https://github.com/cytomining/cytominer-database) or [CytoTable](https://github.com/cytomining/CytoTable).
+For [DeepProfiler](https://cytomining.github.io/DeepProfiler-handbook/docs/00-welcome.html), we include single cell processing tools in [pycytominer.cyto_utils](pycytominer/cyto_utils/).
-From the single cell output, pycytominer performs five steps using a simple API (described below), before passing along data to [cytominer-eval](https://github.com/cytomining/cytominer-eval) for quality and perturbation strength evaluation.
+Next, Pycytominer performs reproducible image-based profiling (**Figure 1B**).
+The Pycytominer API consists of five key steps (**Figure 1C**).
+The outputs generated by Pycytominer are utilized for downstream analysis, which includes machine learning models and statistical testing to derive biological insights.
The best way to communicate with us is through [GitHub Issues](https://github.com/cytomining/pycytominer/issues), where we are able to discuss and troubleshoot topics related to pycytominer.
Please see our [`CONTRIBUTING.md`](https://github.com/cytomining/pycytominer/blob/main/CONTRIBUTING.md) for details about communicating possible bugs, new features, or other information.
@@ -66,6 +70,30 @@ Pycytominer is primarily built on top of [pandas](https://pandas.pydata.org/docs
Pycytominer currently supports [parquet](https://parquet.apache.org/) and compressed text file (e.g. `.csv.gz`) i/o.
+### CellProfiler support
+
+Currently, Pycytominer fully supports data generated by [CellProfiler](https://cellprofiler.org/), adhering defaults to its specific data structure and naming conventions.
+
+CellProfiler-generated image-based profiles typically consist of two main components:
+
+- **Metadata features:** This section contains information about the experiment, such as plate ID, well position, incubation time, perturbation type, and other relevant experimental details. These feature names are prefixed with `Metadata_`, indicating that the data in these columns contain metadata information.
+- **Morphology features:** These are the quantified morphological features prefixed with the default compartments (`Cells_`, `Cytoplasm_`, and `Nuclei_`). Pycytominer also supports non-default compartment names (e.g., `Mito_`).
+
+Note, [`pycytominer.cyto_utils.cells.SingleCells()`](pycytominer/cyto_utils/cells.py) contains code designed to interact with single-cell SQLite files exported from CellProfiler.
+Processing capabilities for SQLite files depends on SQLite file size and your available computational resources (for ex. memory and CPU).
+
+### Handling inputs from other image analysis tools (other than CellProfiler)
+
+Pycytominer also supports processing of raw morphological features from image analysis tools beyond [CellProfiler](https://cellprofiler.org/).
+These tools include [In Carta](https://www.moleculardevices.com/products/cellular-imaging-systems/high-content-analysis/in-carta-image-analysis-software), [Harmony](https://www.revvity.com/product/harmony-5-2-office-revvity-hh17000019#product-overview), and others.
+Using Pycytominer with these tools requires minor modifications to function arguments, and we encourage these users to pay particularly close attention to individual function documentation.
+
+For example, to resolve potential feature issues in the `normalize()` function, you must manually specify the morphological features using the `features` [parameter](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.normalize.normalize).
+The `features` parameter is also available in other key steps, such as [`aggregate`](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.aggregate.aggregate) and [`feature_select`](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.feature_select.feature_select).
+
+If you are using Pycytominer with these other tools, please file [an issue](https://github.com/cytomining/pycytominer/issues) to reach out.
+We'd love to hear from you so that we can learn how to best support broad and multiple use-cases.
+
## API
Pycytominer has five major processing functions:
@@ -97,6 +125,8 @@ Each processing function has unique arguments, see our [documentation](https://p
The default way to use pycytominer is within python scripts, and using pycytominer is simple and fun.
+The example below demonstrates how to perform normalization with a dataset generated by [CellProfiler](https://cellprofiler.org/).
+
```python
# Real world example
import pandas as pd
@@ -135,21 +165,6 @@ And, more specifically than that, image-based profiling readouts from [CellProfi
Therefore, we have included some custom tools in `pycytominer/cyto_utils` that provides other functionality:
-- [Data processing for image-based profiling](#data-processing-for-image-based-profiling)
- - [Installation](#installation)
- - [Frameworks](#frameworks)
- - [API](#api)
- - [Usage](#usage)
- - [Pipeline orchestration](#pipeline-orchestration)
- - [Other functionality](#other-functionality)
- - [CellProfiler CSV collation](#cellprofiler-csv-collation)
- - [Creating a cell locations lookup table](#creating-a-cell-locations-lookup-table)
- - [Generating a GCT file for morpheus](#generating-a-gct-file-for-morpheus)
- - [Citing pycytominer](#citing-pycytominer)
-
-Note, [`pycytominer.cyto_utils.cells.SingleCells()`](pycytominer/cyto_utils/cells.py) contains code to interact with single-cell SQLite files, which are output from CellProfiler.
-Processing capabilities for SQLite files depends on SQLite file size and your available computational resources (for ex. memory and cores).
-
### CellProfiler CSV collation
If running your images on a cluster, unless you have a MySQL or similar large database set up then you will likely end up with lots of different folders from the different cluster runs (often one per well or one per site), each one containing an `Image.csv`, `Nuclei.csv`, etc.
@@ -228,7 +243,7 @@ pycytominer.cyto_utils.write_gct(
)
```
-## Citing pycytominer
+## Citing Pycytominer
If you have used `pycytominer` in your project, please use the citation below.
You can also find the citation in the 'cite this repository' link at the top right under `about` section.
diff --git a/media/legacy_pipeline.png b/media/legacy_pipeline.png
new file mode 100644
index 00000000..c960edfa
Binary files /dev/null and b/media/legacy_pipeline.png differ
diff --git a/media/pipeline.png b/media/pipeline.png
index c960edfa..a164028b 100644
Binary files a/media/pipeline.png and b/media/pipeline.png differ
diff --git a/poetry.lock b/poetry.lock
index de003f88..0b087bbd 100644
--- a/poetry.lock
+++ b/poetry.lock
@@ -1968,6 +1968,64 @@ files = [
{file = "multidict-6.0.5.tar.gz", hash = "sha256:f7e301075edaf50500f0b341543c41194d8df3ae5caf4702f2095f3ca73dd8da"},
]
+[[package]]
+name = "mypy"
+version = "1.11.2"
+description = "Optional static typing for Python"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "mypy-1.11.2-cp310-cp310-macosx_10_9_x86_64.whl", hash = "sha256:d42a6dd818ffce7be66cce644f1dff482f1d97c53ca70908dff0b9ddc120b77a"},
+ {file = "mypy-1.11.2-cp310-cp310-macosx_11_0_arm64.whl", hash = "sha256:801780c56d1cdb896eacd5619a83e427ce436d86a3bdf9112527f24a66618fef"},
+ {file = "mypy-1.11.2-cp310-cp310-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:41ea707d036a5307ac674ea172875f40c9d55c5394f888b168033177fce47383"},
+ {file = "mypy-1.11.2-cp310-cp310-musllinux_1_1_x86_64.whl", hash = "sha256:6e658bd2d20565ea86da7d91331b0eed6d2eee22dc031579e6297f3e12c758c8"},
+ {file = "mypy-1.11.2-cp310-cp310-win_amd64.whl", hash = "sha256:478db5f5036817fe45adb7332d927daa62417159d49783041338921dcf646fc7"},
+ {file = "mypy-1.11.2-cp311-cp311-macosx_10_9_x86_64.whl", hash = "sha256:75746e06d5fa1e91bfd5432448d00d34593b52e7e91a187d981d08d1f33d4385"},
+ {file = "mypy-1.11.2-cp311-cp311-macosx_11_0_arm64.whl", hash = "sha256:a976775ab2256aadc6add633d44f100a2517d2388906ec4f13231fafbb0eccca"},
+ {file = "mypy-1.11.2-cp311-cp311-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:cd953f221ac1379050a8a646585a29574488974f79d8082cedef62744f0a0104"},
+ {file = "mypy-1.11.2-cp311-cp311-musllinux_1_1_x86_64.whl", hash = "sha256:57555a7715c0a34421013144a33d280e73c08df70f3a18a552938587ce9274f4"},
+ {file = "mypy-1.11.2-cp311-cp311-win_amd64.whl", hash = "sha256:36383a4fcbad95f2657642a07ba22ff797de26277158f1cc7bd234821468b1b6"},
+ {file = "mypy-1.11.2-cp312-cp312-macosx_10_9_x86_64.whl", hash = "sha256:e8960dbbbf36906c5c0b7f4fbf2f0c7ffb20f4898e6a879fcf56a41a08b0d318"},
+ {file = "mypy-1.11.2-cp312-cp312-macosx_11_0_arm64.whl", hash = "sha256:06d26c277962f3fb50e13044674aa10553981ae514288cb7d0a738f495550b36"},
+ {file = "mypy-1.11.2-cp312-cp312-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:6e7184632d89d677973a14d00ae4d03214c8bc301ceefcdaf5c474866814c987"},
+ {file = "mypy-1.11.2-cp312-cp312-musllinux_1_1_x86_64.whl", hash = "sha256:3a66169b92452f72117e2da3a576087025449018afc2d8e9bfe5ffab865709ca"},
+ {file = "mypy-1.11.2-cp312-cp312-win_amd64.whl", hash = "sha256:969ea3ef09617aff826885a22ece0ddef69d95852cdad2f60c8bb06bf1f71f70"},
+ {file = "mypy-1.11.2-cp38-cp38-macosx_10_9_x86_64.whl", hash = "sha256:37c7fa6121c1cdfcaac97ce3d3b5588e847aa79b580c1e922bb5d5d2902df19b"},
+ {file = "mypy-1.11.2-cp38-cp38-macosx_11_0_arm64.whl", hash = "sha256:4a8a53bc3ffbd161b5b2a4fff2f0f1e23a33b0168f1c0778ec70e1a3d66deb86"},
+ {file = "mypy-1.11.2-cp38-cp38-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:2ff93107f01968ed834f4256bc1fc4475e2fecf6c661260066a985b52741ddce"},
+ {file = "mypy-1.11.2-cp38-cp38-musllinux_1_1_x86_64.whl", hash = "sha256:edb91dded4df17eae4537668b23f0ff6baf3707683734b6a818d5b9d0c0c31a1"},
+ {file = "mypy-1.11.2-cp38-cp38-win_amd64.whl", hash = "sha256:ee23de8530d99b6db0573c4ef4bd8f39a2a6f9b60655bf7a1357e585a3486f2b"},
+ {file = "mypy-1.11.2-cp39-cp39-macosx_10_9_x86_64.whl", hash = "sha256:801ca29f43d5acce85f8e999b1e431fb479cb02d0e11deb7d2abb56bdaf24fd6"},
+ {file = "mypy-1.11.2-cp39-cp39-macosx_11_0_arm64.whl", hash = "sha256:af8d155170fcf87a2afb55b35dc1a0ac21df4431e7d96717621962e4b9192e70"},
+ {file = "mypy-1.11.2-cp39-cp39-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:f7821776e5c4286b6a13138cc935e2e9b6fde05e081bdebf5cdb2bb97c9df81d"},
+ {file = "mypy-1.11.2-cp39-cp39-musllinux_1_1_x86_64.whl", hash = "sha256:539c570477a96a4e6fb718b8d5c3e0c0eba1f485df13f86d2970c91f0673148d"},
+ {file = "mypy-1.11.2-cp39-cp39-win_amd64.whl", hash = "sha256:3f14cd3d386ac4d05c5a39a51b84387403dadbd936e17cb35882134d4f8f0d24"},
+ {file = "mypy-1.11.2-py3-none-any.whl", hash = "sha256:b499bc07dbdcd3de92b0a8b29fdf592c111276f6a12fe29c30f6c417dd546d12"},
+ {file = "mypy-1.11.2.tar.gz", hash = "sha256:7f9993ad3e0ffdc95c2a14b66dee63729f021968bff8ad911867579c65d13a79"},
+]
+
+[package.dependencies]
+mypy-extensions = ">=1.0.0"
+tomli = {version = ">=1.1.0", markers = "python_version < \"3.11\""}
+typing-extensions = ">=4.6.0"
+
+[package.extras]
+dmypy = ["psutil (>=4.0)"]
+install-types = ["pip"]
+mypyc = ["setuptools (>=50)"]
+reports = ["lxml"]
+
+[[package]]
+name = "mypy-extensions"
+version = "1.0.0"
+description = "Type system extensions for programs checked with the mypy type checker."
+optional = false
+python-versions = ">=3.5"
+files = [
+ {file = "mypy_extensions-1.0.0-py3-none-any.whl", hash = "sha256:4392f6c0eb8a5668a69e23d168ffa70f0be9ccfd32b5cc2d26a34ae5b844552d"},
+ {file = "mypy_extensions-1.0.0.tar.gz", hash = "sha256:75dbf8955dc00442a438fc4d0666508a9a97b6bd41aa2f0ffe9d2f2725af0782"},
+]
+
[[package]]
name = "nbclient"
version = "0.10.0"
@@ -2224,6 +2282,50 @@ sql-other = ["SQLAlchemy (>=1.4.16)"]
test = ["hypothesis (>=6.34.2)", "pytest (>=7.3.2)", "pytest-asyncio (>=0.17.0)", "pytest-xdist (>=2.2.0)"]
xml = ["lxml (>=4.6.3)"]
+[[package]]
+name = "pandas-stubs"
+version = "2.0.2.230605"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "pandas_stubs-2.0.2.230605-py3-none-any.whl", hash = "sha256:39106b602f3cb6dc5f728b84e1b32bde6ecf41ee34ee714c66228009609fbada"},
+ {file = "pandas_stubs-2.0.2.230605.tar.gz", hash = "sha256:624c7bb06d38145a44b61be459ccd19b038e0bf20364a025ecaab78fea65e858"},
+]
+
+[package.dependencies]
+numpy = ">=1.24.3"
+types-pytz = ">=2022.1.1"
+
+[[package]]
+name = "pandas-stubs"
+version = "2.0.3.230814"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "pandas_stubs-2.0.3.230814-py3-none-any.whl", hash = "sha256:4b3dfc027d49779176b7daa031a3405f7b839bcb6e312f4b9f29fea5feec5b4f"},
+ {file = "pandas_stubs-2.0.3.230814.tar.gz", hash = "sha256:1d5cc09e36e3d9f9a1ed9dceae4e03eeb26d1b898dd769996925f784365c8769"},
+]
+
+[package.dependencies]
+types-pytz = ">=2022.1.1"
+
+[[package]]
+name = "pandas-stubs"
+version = "2.2.2.240909"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.10"
+files = [
+ {file = "pandas_stubs-2.2.2.240909-py3-none-any.whl", hash = "sha256:e230f5fa4065f9417804f4d65cd98f86c002efcc07933e8abcd48c3fad9c30a2"},
+ {file = "pandas_stubs-2.2.2.240909.tar.gz", hash = "sha256:3c0951a2c3e45e3475aed9d80b7147ae82f176b9e42e9fb321cfdebf3d411b3d"},
+]
+
+[package.dependencies]
+numpy = ">=1.23.5"
+types-pytz = ">=2022.1.1"
+
[[package]]
name = "pandocfilters"
version = "1.5.1"
@@ -3526,6 +3628,39 @@ files = [
docs = ["myst-parser", "pydata-sphinx-theme", "sphinx"]
test = ["argcomplete (>=3.0.3)", "mypy (>=1.7.0)", "pre-commit", "pytest (>=7.0,<8.1)", "pytest-mock", "pytest-mypy-testing"]
+[[package]]
+name = "types-openpyxl"
+version = "3.1.5.20240918"
+description = "Typing stubs for openpyxl"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "types-openpyxl-3.1.5.20240918.tar.gz", hash = "sha256:22a71a1b601ed8194e356e33e2bebb93dddc47915b410db14ace5a6b7b856955"},
+ {file = "types_openpyxl-3.1.5.20240918-py3-none-any.whl", hash = "sha256:e9bf3c6f7966d347a2514b48f889f272d58e9b22a762244f778a5d66aee2101e"},
+]
+
+[[package]]
+name = "types-pytz"
+version = "2024.2.0.20240913"
+description = "Typing stubs for pytz"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "types-pytz-2024.2.0.20240913.tar.gz", hash = "sha256:4433b5df4a6fc587bbed41716d86a5ba5d832b4378e506f40d34bc9c81df2c24"},
+ {file = "types_pytz-2024.2.0.20240913-py3-none-any.whl", hash = "sha256:a1eebf57ebc6e127a99d2fa2ba0a88d2b173784ef9b3defcc2004ab6855a44df"},
+]
+
+[[package]]
+name = "types-sqlalchemy"
+version = "1.4.53.38"
+description = "Typing stubs for SQLAlchemy"
+optional = false
+python-versions = "*"
+files = [
+ {file = "types-SQLAlchemy-1.4.53.38.tar.gz", hash = "sha256:5bb7463537e04e1aa5a3557eb725930df99226dcfd3c9bf93008025bfe5c169e"},
+ {file = "types_SQLAlchemy-1.4.53.38-py3-none-any.whl", hash = "sha256:7e60e74f823931cc9a9e8adb0a4c05e5533e6708b8a266807893a739faf4eaaa"},
+]
+
[[package]]
name = "typing-extensions"
version = "4.10.0"
diff --git a/pycytominer/aggregate.py b/pycytominer/aggregate.py
index bf8403ba..707f4329 100644
--- a/pycytominer/aggregate.py
+++ b/pycytominer/aggregate.py
@@ -83,7 +83,7 @@ def aggregate(
# Only extract single object column in preparation for count
if compute_object_count:
count_object_df = (
- population_df.loc[:, np.union1d(strata, [object_feature])]
+ population_df.loc[:, list(np.union1d(strata, [object_feature]))]
.groupby(strata)[object_feature]
.count()
.reset_index()
@@ -92,7 +92,9 @@ def aggregate(
if features == "infer":
features = infer_cp_features(population_df)
- population_df = population_df[features]
+
+ # recast as dataframe to protect against scenarios where a series may be returned
+ population_df = pd.DataFrame(population_df[features])
# Fix dtype of input features (they should all be floats!)
population_df = population_df.astype(float)
@@ -101,7 +103,9 @@ def aggregate(
population_df = pd.concat([strata_df, population_df], axis="columns")
# Perform aggregating function
- population_df = population_df.groupby(strata, dropna=False)
+ # Note: type ignore added below to address the change in variable types for
+ # label `population_df`.
+ population_df = population_df.groupby(strata, dropna=False) # type: ignore[assignment]
if operation == "median":
population_df = population_df.median().reset_index()
@@ -118,10 +122,10 @@ def aggregate(
for column in population_df.columns
if column in ["ImageNumber", "ObjectNumber"]
]:
- population_df = population_df.drop([columns_to_drop], axis="columns")
+ population_df = population_df.drop(columns=columns_to_drop, axis="columns")
if output_file is not None:
- output(
+ return output(
df=population_df,
output_filename=output_file,
output_type=output_type,
diff --git a/pycytominer/consensus.py b/pycytominer/consensus.py
index 7ea19714..de0386e5 100644
--- a/pycytominer/consensus.py
+++ b/pycytominer/consensus.py
@@ -35,7 +35,7 @@ def consensus(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
output_file : str, optional
If provided, will write consensus profiles to file. If not specified, will
diff --git a/pycytominer/cyto_utils/DeepProfiler_processing.py b/pycytominer/cyto_utils/DeepProfiler_processing.py
index e33f1036..cc2117d3 100644
--- a/pycytominer/cyto_utils/DeepProfiler_processing.py
+++ b/pycytominer/cyto_utils/DeepProfiler_processing.py
@@ -7,8 +7,9 @@
import pandas as pd
import warnings
-from pycytominer import aggregate, normalize
-from pycytominer.cyto_utils import (
+# use mypy ignores below to avoid duplicate import warnings
+from pycytominer import aggregate, normalize # type: ignore[no-redef]
+from pycytominer.cyto_utils import ( # type: ignore[no-redef]
load_npz_features,
load_npz_locations,
infer_cp_features,
diff --git a/pycytominer/cyto_utils/cell_locations.py b/pycytominer/cyto_utils/cell_locations.py
index 612ae873..5c4edeb7 100644
--- a/pycytominer/cyto_utils/cell_locations.py
+++ b/pycytominer/cyto_utils/cell_locations.py
@@ -106,7 +106,9 @@ def __init__(
"s3", config=botocore.config.Config(signature_version=botocore.UNSIGNED)
)
- def _expanduser(self, obj: Union[str, None]):
+ def _expanduser(
+ self, obj: Union[str, pd.DataFrame, sqlalchemy.engine.Engine, None]
+ ):
"""Expand the user home directory in a path"""
if obj is not None and isinstance(obj, str) and not obj.startswith("s3://"):
return pathlib.Path(obj).expanduser().as_posix()
diff --git a/pycytominer/cyto_utils/cells.py b/pycytominer/cyto_utils/cells.py
index 4e0a80f9..a2f6062f 100644
--- a/pycytominer/cyto_utils/cells.py
+++ b/pycytominer/cyto_utils/cells.py
@@ -714,7 +714,7 @@ def merge_single_cells(
"""
# Load the single cell dataframe by merging on the specific linking columns
- sc_df = ""
+ left_compartment_loaded = False
linking_check_cols = []
merge_suffix_rename = []
for left_compartment in self.compartment_linking_cols:
@@ -737,7 +737,7 @@ def merge_single_cells(
left_compartment
]
- if isinstance(sc_df, str):
+ if not left_compartment_loaded:
sc_df = self.load_compartment(compartment=left_compartment)
if compute_subsample:
@@ -752,6 +752,8 @@ def merge_single_cells(
sc_df, how="left", on=subset_logic_df.columns.tolist()
).reindex(sc_df.columns, axis="columns")
+ left_compartment_loaded = True
+
sc_df = sc_df.merge(
self.load_compartment(compartment=right_compartment),
left_on=[*self.merge_cols, left_link_col],
@@ -804,11 +806,13 @@ def merge_single_cells(
normalize_args["features"] = features
- sc_df = normalize(profiles=sc_df, **normalize_args)
+ # ignore mypy warnings below as these reference root package imports
+ sc_df = normalize(profiles=sc_df, **normalize_args) # type: ignore[operator]
# In case platemap metadata is provided, use pycytominer.annotate for metadata
if platemap is not None:
- sc_df = annotate(
+ # ignore mypy warnings below as these reference root package imports
+ sc_df = annotate( # type: ignore[operator]
profiles=sc_df, platemap=platemap, output_file=None, **kwargs
)
diff --git a/pycytominer/cyto_utils/collate.py b/pycytominer/cyto_utils/collate.py
index a21752fd..4d51b249 100644
--- a/pycytominer/cyto_utils/collate.py
+++ b/pycytominer/cyto_utils/collate.py
@@ -131,7 +131,7 @@ def collate(
with sqlite3.connect(cache_backend_file, isolation_level=None) as connection:
cursor = connection.cursor()
if column:
- if print:
+ if printtoscreen:
print(f"Adding a Metadata_Plate column based on column {column}")
cursor.execute("ALTER TABLE Image ADD COLUMN Metadata_Plate TEXT;")
cursor.execute(f"UPDATE image SET Metadata_Plate ={column};")
diff --git a/pycytominer/cyto_utils/features.py b/pycytominer/cyto_utils/features.py
index 1144a9a4..1d1e4d0d 100644
--- a/pycytominer/cyto_utils/features.py
+++ b/pycytominer/cyto_utils/features.py
@@ -80,7 +80,7 @@ def infer_cp_features(
metadata=False,
image_features=False,
):
- """Given a dataframe, output features that we expect to be Cell Painting features.
+ """Given CellProfiler output data read as a DataFrame, output feature column names as a list.
Parameters
----------
@@ -90,6 +90,8 @@ def infer_cp_features(
Compartments from which Cell Painting features were extracted.
metadata : bool, default False
Whether or not to infer metadata features.
+ If metadata is set to True, find column names that begin with the `Metadata_` prefix.
+ This convention is expected by CellProfiler defaults.
image_features : bool, default False
Whether or not the profiles contain image features.
@@ -115,9 +117,12 @@ def infer_cp_features(
population_df.columns.str.startswith("Metadata_")
].tolist()
- assert ( # noqa: S101
- len(features) > 0
- ), "No CP features found. Are you sure this dataframe is from CellProfiler?"
+ if len(features) == 0:
+ raise ValueError(
+ "No features or metadata found. Pycytominer expects CellProfiler column names by default. "
+ "If you're using non-CellProfiler data, please do not 'infer' features. "
+ "Instead, check if the function has a `features` or `meta_features` parameter, and input column names manually."
+ )
return features
@@ -150,7 +155,9 @@ def drop_outlier_features(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list of str or str, default "infer"
- Features present in the population dataframe. If "infer", then assume Cell Painting features are those that start with "Cells_", "Nuclei_", or "Cytoplasm_"
+ Features present in the population dataframe. If "infer",
+ then assume CellProfiler feature conventions
+ (start with "Cells_", "Nuclei_", or "Cytoplasm_")
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/cyto_utils/modz.py b/pycytominer/cyto_utils/modz.py
index 6e598ed5..6ea4c38c 100644
--- a/pycytominer/cyto_utils/modz.py
+++ b/pycytominer/cyto_utils/modz.py
@@ -98,9 +98,10 @@ def modz(
a string or list of column(s) in the population dataframe that
indicate replicate level information
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
method : str, default "spearman"
indicating which correlation metric to use.
min_weight : float, default 0.01
diff --git a/pycytominer/cyto_utils/output.py b/pycytominer/cyto_utils/output.py
index aa5290a0..62716508 100644
--- a/pycytominer/cyto_utils/output.py
+++ b/pycytominer/cyto_utils/output.py
@@ -2,7 +2,7 @@
Utility function to compress output data
"""
-from typing import Dict, Union, Optional
+from typing import Dict, Union, Optional, Any
import pandas as pd
@@ -12,10 +12,13 @@
def output(
df: pd.DataFrame,
output_filename: str,
- output_type: str = "csv",
+ output_type: Optional[str] = "csv",
sep: str = ",",
float_format: Optional[str] = None,
- compression_options: Union[str, Dict] = {"method": "gzip", "mtime": 1},
+ compression_options: Optional[Union[str, Dict[str, Any]]] = {
+ "method": "gzip",
+ "mtime": 1,
+ },
**kwargs,
):
"""Given an output file and compression options, write file to disk
@@ -79,6 +82,10 @@ def output(
)
"""
+ # ensure a default output type
+ if output_type is None:
+ output_type = "csv"
+
if output_type == "csv":
compression_options = set_compression_method(compression=compression_options)
@@ -98,7 +105,7 @@ def output(
return output_filename
-def set_compression_method(compression: Union[str, Dict]):
+def set_compression_method(compression: Optional[Union[str, Dict]]) -> Dict[str, Any]:
"""Set the compression options
Parameters
diff --git a/pycytominer/cyto_utils/write_gct.py b/pycytominer/cyto_utils/write_gct.py
index 812811bd..1feaab38 100644
--- a/pycytominer/cyto_utils/write_gct.py
+++ b/pycytominer/cyto_utils/write_gct.py
@@ -32,7 +32,7 @@ def write_gct(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
meta_features : list
A list of strings corresponding to metadata column names in the `profiles`
diff --git a/pycytominer/feature_select.py b/pycytominer/feature_select.py
index c1fd87d1..7fc1efab 100644
--- a/pycytominer/feature_select.py
+++ b/pycytominer/feature_select.py
@@ -43,10 +43,10 @@ def feature_select(
----------
profiles : pandas.core.frame.DataFrame or file
DataFrame or file of profiles.
- features : list
+ features : list, default "infer"
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
prefixed with "Cells", "Nuclei", or "Cytoplasm".
image_features: bool, default False
Whether the profiles contain image features.
diff --git a/pycytominer/normalize.py b/pycytominer/normalize.py
index 7a83ca5f..06c55fb6 100644
--- a/pycytominer/normalize.py
+++ b/pycytominer/normalize.py
@@ -34,14 +34,15 @@ def normalize(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
image_features: bool, default False
Whether the profiles contain image features.
meta_features : list
A list of strings corresponding to metadata column names in the `profiles`
DataFrame. All features listed must be found in `profiles`. Defaults to "infer".
- If "infer", then assume metadata features are those prefixed with "Metadata"
+ If "infer", then assume CellProfiler metadata features, identified by
+ column names that begin with the `Metadata_` prefix."
samples : str
The metadata column values to use as a normalization reference. We often use
control samples. The function uses a pd.query() function, so you should
@@ -114,7 +115,7 @@ def normalize(
normalized_df = normalize(
profiles=data_df,
features=["x", "y", "z", "zz"],
- meta_features="infer",
+ meta_features=["Metadata_plate", "Metadata_treatment"],
samples="Metadata_treatment == 'control'",
method="standardize"
)
diff --git a/pycytominer/operations/correlation_threshold.py b/pycytominer/operations/correlation_threshold.py
index 7c4522ba..a888a012 100644
--- a/pycytominer/operations/correlation_threshold.py
+++ b/pycytominer/operations/correlation_threshold.py
@@ -20,9 +20,10 @@ def correlation_threshold(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/operations/get_na_columns.py b/pycytominer/operations/get_na_columns.py
index ad36c377..f288f2cd 100644
--- a/pycytominer/operations/get_na_columns.py
+++ b/pycytominer/operations/get_na_columns.py
@@ -14,9 +14,10 @@ def get_na_columns(population_df, features="infer", samples="all", cutoff=0.05):
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `profiles` DataFrame. All features listed must be found in `profiles`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
@@ -36,8 +37,8 @@ def get_na_columns(population_df, features="infer", samples="all", cutoff=0.05):
if features == "infer":
features = infer_cp_features(population_df)
- else:
- population_df = population_df.loc[:, features]
+
+ population_df = population_df.loc[:, features]
num_rows = population_df.shape[0]
na_prop_df = population_df.isna().sum() / num_rows
diff --git a/pycytominer/operations/noise_removal.py b/pycytominer/operations/noise_removal.py
index aba1a29e..e3e41923 100644
--- a/pycytominer/operations/noise_removal.py
+++ b/pycytominer/operations/noise_removal.py
@@ -22,9 +22,10 @@ def noise_removal(
The list of unique perturbations corresponding to the rows in population_df. For example,
perturb1_well1 and perturb1_well2 would both be "perturb1".
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/operations/variance_threshold.py b/pycytominer/operations/variance_threshold.py
index 67d3b767..72da751d 100644
--- a/pycytominer/operations/variance_threshold.py
+++ b/pycytominer/operations/variance_threshold.py
@@ -18,9 +18,10 @@ def variance_threshold(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pyproject.toml b/pyproject.toml
index 4b5faa17..a00ee920 100644
--- a/pyproject.toml
+++ b/pyproject.toml
@@ -73,6 +73,13 @@ pytest-cov = "^4.1.0"
pre-commit = ">=3.3.2"
commitizen = "^3.12.0"
ruff = "^0.3.4"
+mypy = "^1.11.2"
+types-openpyxl = "^3.1.5.20240918"
+pandas-stubs = [
+ {version = "<2.2.2.240909", python = "<3.10"},
+ {version = "^2.2.2.240909", python = ">=3.10"}
+]
+types-sqlalchemy = "^1.4.53.38"
[tool.poetry.group.docs]
optional = true
@@ -177,6 +184,29 @@ preview = true
[tool.pytest.ini_options]
testpaths = "tests"
+[tool.mypy]
+exclude = [
+ # ignore notebook-based walkthroughs
+ "walkthroughs",
+ # ignore tests dir
+ "tests"
+]
+
+
+[[tool.mypy.overrides]]
+# ignore missing import errors for the following
+# packages which do not have standard type stubs
+# or experience issues with imported types.
+module = [
+ "sklearn.*",
+ "scipy.*",
+ "cytominer_database.*",
+ "fire.*",
+ "boto3.*",
+ "botocore.*"
+]
+ignore_missing_imports = true
+
[build-system]
requires = ["poetry-core>=1.7.0", "poetry-dynamic-versioning>=1.1.0"]
build-backend = "poetry_dynamic_versioning.backend"
diff --git a/tests/test_aggregate.py b/tests/test_aggregate.py
index d1a08889..7765ec31 100644
--- a/tests/test_aggregate.py
+++ b/tests/test_aggregate.py
@@ -64,6 +64,19 @@ def test_aggregate_median_allvar():
assert aggregate_result.equals(expected_result)
+ # Test that ImageNumber column would be dropped before return
+ data_df_with_imagenumber = data_df.copy()
+ data_df_with_imagenumber["ImageNumber"] = "1"
+
+ aggregate_result = aggregate(
+ population_df=data_df_with_imagenumber,
+ strata=["g"],
+ features="infer",
+ operation="median",
+ )
+
+ assert aggregate_result.equals(expected_result)
+
# Test output
aggregate(
population_df=data_df,
diff --git a/tests/test_cyto_utils/test_feature_infer.py b/tests/test_cyto_utils/test_feature_infer.py
index df839858..9c8e8e2a 100644
--- a/tests/test_cyto_utils/test_feature_infer.py
+++ b/tests/test_cyto_utils/test_feature_infer.py
@@ -39,10 +39,10 @@ def test_feature_infer():
def test_feature_infer_nocp():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
infer_cp_features(population_df=non_cp_data_df)
- assert "No CP features found." in str(nocp.value)
+ assert "No features or metadata found." in str(nocp.value)
def test_metadata_feature_infer():
diff --git a/tests/test_cyto_utils/test_output.py b/tests/test_cyto_utils/test_output.py
index 72d5a9cf..0aeefd06 100644
--- a/tests/test_cyto_utils/test_output.py
+++ b/tests/test_cyto_utils/test_output.py
@@ -56,6 +56,20 @@ def test_output_default():
result, DATA_DF, check_names=False, check_exact=False, atol=1e-3
)
+ # test with an output_type of None
+ output_result = output(
+ df=DATA_DF,
+ output_filename=output_filename,
+ compression_options=TEST_COMPRESSION_OPTIONS,
+ float_format=None,
+ output_type=None,
+ )
+ result = pd.read_csv(output_result)
+
+ pd.testing.assert_frame_equal(
+ result, DATA_DF, check_names=False, check_exact=False, atol=1e-3
+ )
+
def test_output_tsv():
# Test input filename of writing a tab separated file
diff --git a/tests/test_operations/test_correlation_threshold.py b/tests/test_operations/test_correlation_threshold.py
index 2ca393cc..9845b99f 100644
--- a/tests/test_operations/test_correlation_threshold.py
+++ b/tests/test_operations/test_correlation_threshold.py
@@ -75,7 +75,7 @@ def test_correlation_threshold_samples():
def test_correlation_threshold_featureinfer():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
correlation_threshold_result = correlation_threshold(
population_df=data_df,
features="infer",
@@ -84,7 +84,7 @@ def test_correlation_threshold_featureinfer():
method="pearson",
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
data_cp_df = data_df.copy()
data_cp_df.columns = [f"Cells_{x}" for x in data_df.columns]
diff --git a/tests/test_operations/test_get_na_columns.py b/tests/test_operations/test_get_na_columns.py
index 9c8bd557..48b5ab71 100644
--- a/tests/test_operations/test_get_na_columns.py
+++ b/tests/test_operations/test_get_na_columns.py
@@ -67,9 +67,9 @@ def test_get_na_columns_sample():
def test_get_na_columns_featureinfer():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
get_na_columns(
population_df=data_df, samples="all", features="infer", cutoff=0.1
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
diff --git a/tests/test_operations/test_variance_threshold.py b/tests/test_operations/test_variance_threshold.py
index a1a19764..1d7cd481 100644
--- a/tests/test_operations/test_variance_threshold.py
+++ b/tests/test_operations/test_variance_threshold.py
@@ -102,12 +102,12 @@ def test_variance_threshold():
def test_variance_threshold_featureinfer():
unique_cut = 0.01
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
excluded_features = variance_threshold(
population_df=data_unique_test_df, features="infer", unique_cut=unique_cut
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
data_cp_df = data_unique_test_df.copy()
data_cp_df.columns = [f"Cells_{x}" for x in data_unique_test_df.columns]
+
+
+> Figure 1. The standard image-based profiling experiment and the role of Pycytominer. (A) In the experimental phase, a scientist plates cells, often perturbing them with chemical or genetic agents and performs microscopy imaging. In image analysis, using CellProfiler for example, a scientist applies several data processing steps to generate image-based profiles. In addition, scientists can apply a more flexible approach by using deep learning models, such as DeepProfiler, to generate image-based profiles. (B) Pycytominer performs image-based profiling to process morphology features and make them ready for downstream analyses. (C) Pycytominer performs five fundamental functions, each implemented with a simple and intuitive API. Each function enables a user to implement various methods for executing operations.
[Click here for high resolution pipeline image](https://github.com/cytomining/pycytominer/blob/main/media/pipeline.png)
-Image data flow from a microscope to cell segmentation and feature extraction tools (e.g. CellProfiler or DeepProfiler).
+Image data flow from a microscope to cell segmentation and feature extraction tools (e.g. [CellProfiler](https://cellprofiler.org/) or [DeepProfiler](https://cytomining.github.io/DeepProfiler-handbook/docs/00-welcome.html)) (**Figure 1A**).
From here, additional single cell processing tools curate the single cell readouts into a form manageable for pycytominer input.
-For CellProfiler, we use [cytominer-database](https://github.com/cytomining/cytominer-database) or [CytoTable](https://github.com/cytomining/CytoTable).
-For DeepProfiler, we include single cell processing tools in [pycytominer.cyto_utils](pycytominer/cyto_utils/).
+For [CellProfiler](https://cellprofiler.org/), we use [cytominer-database](https://github.com/cytomining/cytominer-database) or [CytoTable](https://github.com/cytomining/CytoTable).
+For [DeepProfiler](https://cytomining.github.io/DeepProfiler-handbook/docs/00-welcome.html), we include single cell processing tools in [pycytominer.cyto_utils](pycytominer/cyto_utils/).
-From the single cell output, pycytominer performs five steps using a simple API (described below), before passing along data to [cytominer-eval](https://github.com/cytomining/cytominer-eval) for quality and perturbation strength evaluation.
+Next, Pycytominer performs reproducible image-based profiling (**Figure 1B**).
+The Pycytominer API consists of five key steps (**Figure 1C**).
+The outputs generated by Pycytominer are utilized for downstream analysis, which includes machine learning models and statistical testing to derive biological insights.
The best way to communicate with us is through [GitHub Issues](https://github.com/cytomining/pycytominer/issues), where we are able to discuss and troubleshoot topics related to pycytominer.
Please see our [`CONTRIBUTING.md`](https://github.com/cytomining/pycytominer/blob/main/CONTRIBUTING.md) for details about communicating possible bugs, new features, or other information.
@@ -66,6 +70,30 @@ Pycytominer is primarily built on top of [pandas](https://pandas.pydata.org/docs
Pycytominer currently supports [parquet](https://parquet.apache.org/) and compressed text file (e.g. `.csv.gz`) i/o.
+### CellProfiler support
+
+Currently, Pycytominer fully supports data generated by [CellProfiler](https://cellprofiler.org/), adhering defaults to its specific data structure and naming conventions.
+
+CellProfiler-generated image-based profiles typically consist of two main components:
+
+- **Metadata features:** This section contains information about the experiment, such as plate ID, well position, incubation time, perturbation type, and other relevant experimental details. These feature names are prefixed with `Metadata_`, indicating that the data in these columns contain metadata information.
+- **Morphology features:** These are the quantified morphological features prefixed with the default compartments (`Cells_`, `Cytoplasm_`, and `Nuclei_`). Pycytominer also supports non-default compartment names (e.g., `Mito_`).
+
+Note, [`pycytominer.cyto_utils.cells.SingleCells()`](pycytominer/cyto_utils/cells.py) contains code designed to interact with single-cell SQLite files exported from CellProfiler.
+Processing capabilities for SQLite files depends on SQLite file size and your available computational resources (for ex. memory and CPU).
+
+### Handling inputs from other image analysis tools (other than CellProfiler)
+
+Pycytominer also supports processing of raw morphological features from image analysis tools beyond [CellProfiler](https://cellprofiler.org/).
+These tools include [In Carta](https://www.moleculardevices.com/products/cellular-imaging-systems/high-content-analysis/in-carta-image-analysis-software), [Harmony](https://www.revvity.com/product/harmony-5-2-office-revvity-hh17000019#product-overview), and others.
+Using Pycytominer with these tools requires minor modifications to function arguments, and we encourage these users to pay particularly close attention to individual function documentation.
+
+For example, to resolve potential feature issues in the `normalize()` function, you must manually specify the morphological features using the `features` [parameter](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.normalize.normalize).
+The `features` parameter is also available in other key steps, such as [`aggregate`](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.aggregate.aggregate) and [`feature_select`](https://pycytominer.readthedocs.io/en/latest/pycytominer.html#pycytominer.feature_select.feature_select).
+
+If you are using Pycytominer with these other tools, please file [an issue](https://github.com/cytomining/pycytominer/issues) to reach out.
+We'd love to hear from you so that we can learn how to best support broad and multiple use-cases.
+
## API
Pycytominer has five major processing functions:
@@ -97,6 +125,8 @@ Each processing function has unique arguments, see our [documentation](https://p
The default way to use pycytominer is within python scripts, and using pycytominer is simple and fun.
+The example below demonstrates how to perform normalization with a dataset generated by [CellProfiler](https://cellprofiler.org/).
+
```python
# Real world example
import pandas as pd
@@ -135,21 +165,6 @@ And, more specifically than that, image-based profiling readouts from [CellProfi
Therefore, we have included some custom tools in `pycytominer/cyto_utils` that provides other functionality:
-- [Data processing for image-based profiling](#data-processing-for-image-based-profiling)
- - [Installation](#installation)
- - [Frameworks](#frameworks)
- - [API](#api)
- - [Usage](#usage)
- - [Pipeline orchestration](#pipeline-orchestration)
- - [Other functionality](#other-functionality)
- - [CellProfiler CSV collation](#cellprofiler-csv-collation)
- - [Creating a cell locations lookup table](#creating-a-cell-locations-lookup-table)
- - [Generating a GCT file for morpheus](#generating-a-gct-file-for-morpheus)
- - [Citing pycytominer](#citing-pycytominer)
-
-Note, [`pycytominer.cyto_utils.cells.SingleCells()`](pycytominer/cyto_utils/cells.py) contains code to interact with single-cell SQLite files, which are output from CellProfiler.
-Processing capabilities for SQLite files depends on SQLite file size and your available computational resources (for ex. memory and cores).
-
### CellProfiler CSV collation
If running your images on a cluster, unless you have a MySQL or similar large database set up then you will likely end up with lots of different folders from the different cluster runs (often one per well or one per site), each one containing an `Image.csv`, `Nuclei.csv`, etc.
@@ -228,7 +243,7 @@ pycytominer.cyto_utils.write_gct(
)
```
-## Citing pycytominer
+## Citing Pycytominer
If you have used `pycytominer` in your project, please use the citation below.
You can also find the citation in the 'cite this repository' link at the top right under `about` section.
diff --git a/media/legacy_pipeline.png b/media/legacy_pipeline.png
new file mode 100644
index 00000000..c960edfa
Binary files /dev/null and b/media/legacy_pipeline.png differ
diff --git a/media/pipeline.png b/media/pipeline.png
index c960edfa..a164028b 100644
Binary files a/media/pipeline.png and b/media/pipeline.png differ
diff --git a/poetry.lock b/poetry.lock
index de003f88..0b087bbd 100644
--- a/poetry.lock
+++ b/poetry.lock
@@ -1968,6 +1968,64 @@ files = [
{file = "multidict-6.0.5.tar.gz", hash = "sha256:f7e301075edaf50500f0b341543c41194d8df3ae5caf4702f2095f3ca73dd8da"},
]
+[[package]]
+name = "mypy"
+version = "1.11.2"
+description = "Optional static typing for Python"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "mypy-1.11.2-cp310-cp310-macosx_10_9_x86_64.whl", hash = "sha256:d42a6dd818ffce7be66cce644f1dff482f1d97c53ca70908dff0b9ddc120b77a"},
+ {file = "mypy-1.11.2-cp310-cp310-macosx_11_0_arm64.whl", hash = "sha256:801780c56d1cdb896eacd5619a83e427ce436d86a3bdf9112527f24a66618fef"},
+ {file = "mypy-1.11.2-cp310-cp310-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:41ea707d036a5307ac674ea172875f40c9d55c5394f888b168033177fce47383"},
+ {file = "mypy-1.11.2-cp310-cp310-musllinux_1_1_x86_64.whl", hash = "sha256:6e658bd2d20565ea86da7d91331b0eed6d2eee22dc031579e6297f3e12c758c8"},
+ {file = "mypy-1.11.2-cp310-cp310-win_amd64.whl", hash = "sha256:478db5f5036817fe45adb7332d927daa62417159d49783041338921dcf646fc7"},
+ {file = "mypy-1.11.2-cp311-cp311-macosx_10_9_x86_64.whl", hash = "sha256:75746e06d5fa1e91bfd5432448d00d34593b52e7e91a187d981d08d1f33d4385"},
+ {file = "mypy-1.11.2-cp311-cp311-macosx_11_0_arm64.whl", hash = "sha256:a976775ab2256aadc6add633d44f100a2517d2388906ec4f13231fafbb0eccca"},
+ {file = "mypy-1.11.2-cp311-cp311-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:cd953f221ac1379050a8a646585a29574488974f79d8082cedef62744f0a0104"},
+ {file = "mypy-1.11.2-cp311-cp311-musllinux_1_1_x86_64.whl", hash = "sha256:57555a7715c0a34421013144a33d280e73c08df70f3a18a552938587ce9274f4"},
+ {file = "mypy-1.11.2-cp311-cp311-win_amd64.whl", hash = "sha256:36383a4fcbad95f2657642a07ba22ff797de26277158f1cc7bd234821468b1b6"},
+ {file = "mypy-1.11.2-cp312-cp312-macosx_10_9_x86_64.whl", hash = "sha256:e8960dbbbf36906c5c0b7f4fbf2f0c7ffb20f4898e6a879fcf56a41a08b0d318"},
+ {file = "mypy-1.11.2-cp312-cp312-macosx_11_0_arm64.whl", hash = "sha256:06d26c277962f3fb50e13044674aa10553981ae514288cb7d0a738f495550b36"},
+ {file = "mypy-1.11.2-cp312-cp312-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:6e7184632d89d677973a14d00ae4d03214c8bc301ceefcdaf5c474866814c987"},
+ {file = "mypy-1.11.2-cp312-cp312-musllinux_1_1_x86_64.whl", hash = "sha256:3a66169b92452f72117e2da3a576087025449018afc2d8e9bfe5ffab865709ca"},
+ {file = "mypy-1.11.2-cp312-cp312-win_amd64.whl", hash = "sha256:969ea3ef09617aff826885a22ece0ddef69d95852cdad2f60c8bb06bf1f71f70"},
+ {file = "mypy-1.11.2-cp38-cp38-macosx_10_9_x86_64.whl", hash = "sha256:37c7fa6121c1cdfcaac97ce3d3b5588e847aa79b580c1e922bb5d5d2902df19b"},
+ {file = "mypy-1.11.2-cp38-cp38-macosx_11_0_arm64.whl", hash = "sha256:4a8a53bc3ffbd161b5b2a4fff2f0f1e23a33b0168f1c0778ec70e1a3d66deb86"},
+ {file = "mypy-1.11.2-cp38-cp38-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:2ff93107f01968ed834f4256bc1fc4475e2fecf6c661260066a985b52741ddce"},
+ {file = "mypy-1.11.2-cp38-cp38-musllinux_1_1_x86_64.whl", hash = "sha256:edb91dded4df17eae4537668b23f0ff6baf3707683734b6a818d5b9d0c0c31a1"},
+ {file = "mypy-1.11.2-cp38-cp38-win_amd64.whl", hash = "sha256:ee23de8530d99b6db0573c4ef4bd8f39a2a6f9b60655bf7a1357e585a3486f2b"},
+ {file = "mypy-1.11.2-cp39-cp39-macosx_10_9_x86_64.whl", hash = "sha256:801ca29f43d5acce85f8e999b1e431fb479cb02d0e11deb7d2abb56bdaf24fd6"},
+ {file = "mypy-1.11.2-cp39-cp39-macosx_11_0_arm64.whl", hash = "sha256:af8d155170fcf87a2afb55b35dc1a0ac21df4431e7d96717621962e4b9192e70"},
+ {file = "mypy-1.11.2-cp39-cp39-manylinux_2_17_x86_64.manylinux2014_x86_64.manylinux_2_28_x86_64.whl", hash = "sha256:f7821776e5c4286b6a13138cc935e2e9b6fde05e081bdebf5cdb2bb97c9df81d"},
+ {file = "mypy-1.11.2-cp39-cp39-musllinux_1_1_x86_64.whl", hash = "sha256:539c570477a96a4e6fb718b8d5c3e0c0eba1f485df13f86d2970c91f0673148d"},
+ {file = "mypy-1.11.2-cp39-cp39-win_amd64.whl", hash = "sha256:3f14cd3d386ac4d05c5a39a51b84387403dadbd936e17cb35882134d4f8f0d24"},
+ {file = "mypy-1.11.2-py3-none-any.whl", hash = "sha256:b499bc07dbdcd3de92b0a8b29fdf592c111276f6a12fe29c30f6c417dd546d12"},
+ {file = "mypy-1.11.2.tar.gz", hash = "sha256:7f9993ad3e0ffdc95c2a14b66dee63729f021968bff8ad911867579c65d13a79"},
+]
+
+[package.dependencies]
+mypy-extensions = ">=1.0.0"
+tomli = {version = ">=1.1.0", markers = "python_version < \"3.11\""}
+typing-extensions = ">=4.6.0"
+
+[package.extras]
+dmypy = ["psutil (>=4.0)"]
+install-types = ["pip"]
+mypyc = ["setuptools (>=50)"]
+reports = ["lxml"]
+
+[[package]]
+name = "mypy-extensions"
+version = "1.0.0"
+description = "Type system extensions for programs checked with the mypy type checker."
+optional = false
+python-versions = ">=3.5"
+files = [
+ {file = "mypy_extensions-1.0.0-py3-none-any.whl", hash = "sha256:4392f6c0eb8a5668a69e23d168ffa70f0be9ccfd32b5cc2d26a34ae5b844552d"},
+ {file = "mypy_extensions-1.0.0.tar.gz", hash = "sha256:75dbf8955dc00442a438fc4d0666508a9a97b6bd41aa2f0ffe9d2f2725af0782"},
+]
+
[[package]]
name = "nbclient"
version = "0.10.0"
@@ -2224,6 +2282,50 @@ sql-other = ["SQLAlchemy (>=1.4.16)"]
test = ["hypothesis (>=6.34.2)", "pytest (>=7.3.2)", "pytest-asyncio (>=0.17.0)", "pytest-xdist (>=2.2.0)"]
xml = ["lxml (>=4.6.3)"]
+[[package]]
+name = "pandas-stubs"
+version = "2.0.2.230605"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "pandas_stubs-2.0.2.230605-py3-none-any.whl", hash = "sha256:39106b602f3cb6dc5f728b84e1b32bde6ecf41ee34ee714c66228009609fbada"},
+ {file = "pandas_stubs-2.0.2.230605.tar.gz", hash = "sha256:624c7bb06d38145a44b61be459ccd19b038e0bf20364a025ecaab78fea65e858"},
+]
+
+[package.dependencies]
+numpy = ">=1.24.3"
+types-pytz = ">=2022.1.1"
+
+[[package]]
+name = "pandas-stubs"
+version = "2.0.3.230814"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "pandas_stubs-2.0.3.230814-py3-none-any.whl", hash = "sha256:4b3dfc027d49779176b7daa031a3405f7b839bcb6e312f4b9f29fea5feec5b4f"},
+ {file = "pandas_stubs-2.0.3.230814.tar.gz", hash = "sha256:1d5cc09e36e3d9f9a1ed9dceae4e03eeb26d1b898dd769996925f784365c8769"},
+]
+
+[package.dependencies]
+types-pytz = ">=2022.1.1"
+
+[[package]]
+name = "pandas-stubs"
+version = "2.2.2.240909"
+description = "Type annotations for pandas"
+optional = false
+python-versions = ">=3.10"
+files = [
+ {file = "pandas_stubs-2.2.2.240909-py3-none-any.whl", hash = "sha256:e230f5fa4065f9417804f4d65cd98f86c002efcc07933e8abcd48c3fad9c30a2"},
+ {file = "pandas_stubs-2.2.2.240909.tar.gz", hash = "sha256:3c0951a2c3e45e3475aed9d80b7147ae82f176b9e42e9fb321cfdebf3d411b3d"},
+]
+
+[package.dependencies]
+numpy = ">=1.23.5"
+types-pytz = ">=2022.1.1"
+
[[package]]
name = "pandocfilters"
version = "1.5.1"
@@ -3526,6 +3628,39 @@ files = [
docs = ["myst-parser", "pydata-sphinx-theme", "sphinx"]
test = ["argcomplete (>=3.0.3)", "mypy (>=1.7.0)", "pre-commit", "pytest (>=7.0,<8.1)", "pytest-mock", "pytest-mypy-testing"]
+[[package]]
+name = "types-openpyxl"
+version = "3.1.5.20240918"
+description = "Typing stubs for openpyxl"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "types-openpyxl-3.1.5.20240918.tar.gz", hash = "sha256:22a71a1b601ed8194e356e33e2bebb93dddc47915b410db14ace5a6b7b856955"},
+ {file = "types_openpyxl-3.1.5.20240918-py3-none-any.whl", hash = "sha256:e9bf3c6f7966d347a2514b48f889f272d58e9b22a762244f778a5d66aee2101e"},
+]
+
+[[package]]
+name = "types-pytz"
+version = "2024.2.0.20240913"
+description = "Typing stubs for pytz"
+optional = false
+python-versions = ">=3.8"
+files = [
+ {file = "types-pytz-2024.2.0.20240913.tar.gz", hash = "sha256:4433b5df4a6fc587bbed41716d86a5ba5d832b4378e506f40d34bc9c81df2c24"},
+ {file = "types_pytz-2024.2.0.20240913-py3-none-any.whl", hash = "sha256:a1eebf57ebc6e127a99d2fa2ba0a88d2b173784ef9b3defcc2004ab6855a44df"},
+]
+
+[[package]]
+name = "types-sqlalchemy"
+version = "1.4.53.38"
+description = "Typing stubs for SQLAlchemy"
+optional = false
+python-versions = "*"
+files = [
+ {file = "types-SQLAlchemy-1.4.53.38.tar.gz", hash = "sha256:5bb7463537e04e1aa5a3557eb725930df99226dcfd3c9bf93008025bfe5c169e"},
+ {file = "types_SQLAlchemy-1.4.53.38-py3-none-any.whl", hash = "sha256:7e60e74f823931cc9a9e8adb0a4c05e5533e6708b8a266807893a739faf4eaaa"},
+]
+
[[package]]
name = "typing-extensions"
version = "4.10.0"
diff --git a/pycytominer/aggregate.py b/pycytominer/aggregate.py
index bf8403ba..707f4329 100644
--- a/pycytominer/aggregate.py
+++ b/pycytominer/aggregate.py
@@ -83,7 +83,7 @@ def aggregate(
# Only extract single object column in preparation for count
if compute_object_count:
count_object_df = (
- population_df.loc[:, np.union1d(strata, [object_feature])]
+ population_df.loc[:, list(np.union1d(strata, [object_feature]))]
.groupby(strata)[object_feature]
.count()
.reset_index()
@@ -92,7 +92,9 @@ def aggregate(
if features == "infer":
features = infer_cp_features(population_df)
- population_df = population_df[features]
+
+ # recast as dataframe to protect against scenarios where a series may be returned
+ population_df = pd.DataFrame(population_df[features])
# Fix dtype of input features (they should all be floats!)
population_df = population_df.astype(float)
@@ -101,7 +103,9 @@ def aggregate(
population_df = pd.concat([strata_df, population_df], axis="columns")
# Perform aggregating function
- population_df = population_df.groupby(strata, dropna=False)
+ # Note: type ignore added below to address the change in variable types for
+ # label `population_df`.
+ population_df = population_df.groupby(strata, dropna=False) # type: ignore[assignment]
if operation == "median":
population_df = population_df.median().reset_index()
@@ -118,10 +122,10 @@ def aggregate(
for column in population_df.columns
if column in ["ImageNumber", "ObjectNumber"]
]:
- population_df = population_df.drop([columns_to_drop], axis="columns")
+ population_df = population_df.drop(columns=columns_to_drop, axis="columns")
if output_file is not None:
- output(
+ return output(
df=population_df,
output_filename=output_file,
output_type=output_type,
diff --git a/pycytominer/consensus.py b/pycytominer/consensus.py
index 7ea19714..de0386e5 100644
--- a/pycytominer/consensus.py
+++ b/pycytominer/consensus.py
@@ -35,7 +35,7 @@ def consensus(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
output_file : str, optional
If provided, will write consensus profiles to file. If not specified, will
diff --git a/pycytominer/cyto_utils/DeepProfiler_processing.py b/pycytominer/cyto_utils/DeepProfiler_processing.py
index e33f1036..cc2117d3 100644
--- a/pycytominer/cyto_utils/DeepProfiler_processing.py
+++ b/pycytominer/cyto_utils/DeepProfiler_processing.py
@@ -7,8 +7,9 @@
import pandas as pd
import warnings
-from pycytominer import aggregate, normalize
-from pycytominer.cyto_utils import (
+# use mypy ignores below to avoid duplicate import warnings
+from pycytominer import aggregate, normalize # type: ignore[no-redef]
+from pycytominer.cyto_utils import ( # type: ignore[no-redef]
load_npz_features,
load_npz_locations,
infer_cp_features,
diff --git a/pycytominer/cyto_utils/cell_locations.py b/pycytominer/cyto_utils/cell_locations.py
index 612ae873..5c4edeb7 100644
--- a/pycytominer/cyto_utils/cell_locations.py
+++ b/pycytominer/cyto_utils/cell_locations.py
@@ -106,7 +106,9 @@ def __init__(
"s3", config=botocore.config.Config(signature_version=botocore.UNSIGNED)
)
- def _expanduser(self, obj: Union[str, None]):
+ def _expanduser(
+ self, obj: Union[str, pd.DataFrame, sqlalchemy.engine.Engine, None]
+ ):
"""Expand the user home directory in a path"""
if obj is not None and isinstance(obj, str) and not obj.startswith("s3://"):
return pathlib.Path(obj).expanduser().as_posix()
diff --git a/pycytominer/cyto_utils/cells.py b/pycytominer/cyto_utils/cells.py
index 4e0a80f9..a2f6062f 100644
--- a/pycytominer/cyto_utils/cells.py
+++ b/pycytominer/cyto_utils/cells.py
@@ -714,7 +714,7 @@ def merge_single_cells(
"""
# Load the single cell dataframe by merging on the specific linking columns
- sc_df = ""
+ left_compartment_loaded = False
linking_check_cols = []
merge_suffix_rename = []
for left_compartment in self.compartment_linking_cols:
@@ -737,7 +737,7 @@ def merge_single_cells(
left_compartment
]
- if isinstance(sc_df, str):
+ if not left_compartment_loaded:
sc_df = self.load_compartment(compartment=left_compartment)
if compute_subsample:
@@ -752,6 +752,8 @@ def merge_single_cells(
sc_df, how="left", on=subset_logic_df.columns.tolist()
).reindex(sc_df.columns, axis="columns")
+ left_compartment_loaded = True
+
sc_df = sc_df.merge(
self.load_compartment(compartment=right_compartment),
left_on=[*self.merge_cols, left_link_col],
@@ -804,11 +806,13 @@ def merge_single_cells(
normalize_args["features"] = features
- sc_df = normalize(profiles=sc_df, **normalize_args)
+ # ignore mypy warnings below as these reference root package imports
+ sc_df = normalize(profiles=sc_df, **normalize_args) # type: ignore[operator]
# In case platemap metadata is provided, use pycytominer.annotate for metadata
if platemap is not None:
- sc_df = annotate(
+ # ignore mypy warnings below as these reference root package imports
+ sc_df = annotate( # type: ignore[operator]
profiles=sc_df, platemap=platemap, output_file=None, **kwargs
)
diff --git a/pycytominer/cyto_utils/collate.py b/pycytominer/cyto_utils/collate.py
index a21752fd..4d51b249 100644
--- a/pycytominer/cyto_utils/collate.py
+++ b/pycytominer/cyto_utils/collate.py
@@ -131,7 +131,7 @@ def collate(
with sqlite3.connect(cache_backend_file, isolation_level=None) as connection:
cursor = connection.cursor()
if column:
- if print:
+ if printtoscreen:
print(f"Adding a Metadata_Plate column based on column {column}")
cursor.execute("ALTER TABLE Image ADD COLUMN Metadata_Plate TEXT;")
cursor.execute(f"UPDATE image SET Metadata_Plate ={column};")
diff --git a/pycytominer/cyto_utils/features.py b/pycytominer/cyto_utils/features.py
index 1144a9a4..1d1e4d0d 100644
--- a/pycytominer/cyto_utils/features.py
+++ b/pycytominer/cyto_utils/features.py
@@ -80,7 +80,7 @@ def infer_cp_features(
metadata=False,
image_features=False,
):
- """Given a dataframe, output features that we expect to be Cell Painting features.
+ """Given CellProfiler output data read as a DataFrame, output feature column names as a list.
Parameters
----------
@@ -90,6 +90,8 @@ def infer_cp_features(
Compartments from which Cell Painting features were extracted.
metadata : bool, default False
Whether or not to infer metadata features.
+ If metadata is set to True, find column names that begin with the `Metadata_` prefix.
+ This convention is expected by CellProfiler defaults.
image_features : bool, default False
Whether or not the profiles contain image features.
@@ -115,9 +117,12 @@ def infer_cp_features(
population_df.columns.str.startswith("Metadata_")
].tolist()
- assert ( # noqa: S101
- len(features) > 0
- ), "No CP features found. Are you sure this dataframe is from CellProfiler?"
+ if len(features) == 0:
+ raise ValueError(
+ "No features or metadata found. Pycytominer expects CellProfiler column names by default. "
+ "If you're using non-CellProfiler data, please do not 'infer' features. "
+ "Instead, check if the function has a `features` or `meta_features` parameter, and input column names manually."
+ )
return features
@@ -150,7 +155,9 @@ def drop_outlier_features(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list of str or str, default "infer"
- Features present in the population dataframe. If "infer", then assume Cell Painting features are those that start with "Cells_", "Nuclei_", or "Cytoplasm_"
+ Features present in the population dataframe. If "infer",
+ then assume CellProfiler feature conventions
+ (start with "Cells_", "Nuclei_", or "Cytoplasm_")
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/cyto_utils/modz.py b/pycytominer/cyto_utils/modz.py
index 6e598ed5..6ea4c38c 100644
--- a/pycytominer/cyto_utils/modz.py
+++ b/pycytominer/cyto_utils/modz.py
@@ -98,9 +98,10 @@ def modz(
a string or list of column(s) in the population dataframe that
indicate replicate level information
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
method : str, default "spearman"
indicating which correlation metric to use.
min_weight : float, default 0.01
diff --git a/pycytominer/cyto_utils/output.py b/pycytominer/cyto_utils/output.py
index aa5290a0..62716508 100644
--- a/pycytominer/cyto_utils/output.py
+++ b/pycytominer/cyto_utils/output.py
@@ -2,7 +2,7 @@
Utility function to compress output data
"""
-from typing import Dict, Union, Optional
+from typing import Dict, Union, Optional, Any
import pandas as pd
@@ -12,10 +12,13 @@
def output(
df: pd.DataFrame,
output_filename: str,
- output_type: str = "csv",
+ output_type: Optional[str] = "csv",
sep: str = ",",
float_format: Optional[str] = None,
- compression_options: Union[str, Dict] = {"method": "gzip", "mtime": 1},
+ compression_options: Optional[Union[str, Dict[str, Any]]] = {
+ "method": "gzip",
+ "mtime": 1,
+ },
**kwargs,
):
"""Given an output file and compression options, write file to disk
@@ -79,6 +82,10 @@ def output(
)
"""
+ # ensure a default output type
+ if output_type is None:
+ output_type = "csv"
+
if output_type == "csv":
compression_options = set_compression_method(compression=compression_options)
@@ -98,7 +105,7 @@ def output(
return output_filename
-def set_compression_method(compression: Union[str, Dict]):
+def set_compression_method(compression: Optional[Union[str, Dict]]) -> Dict[str, Any]:
"""Set the compression options
Parameters
diff --git a/pycytominer/cyto_utils/write_gct.py b/pycytominer/cyto_utils/write_gct.py
index 812811bd..1feaab38 100644
--- a/pycytominer/cyto_utils/write_gct.py
+++ b/pycytominer/cyto_utils/write_gct.py
@@ -32,7 +32,7 @@ def write_gct(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
meta_features : list
A list of strings corresponding to metadata column names in the `profiles`
diff --git a/pycytominer/feature_select.py b/pycytominer/feature_select.py
index c1fd87d1..7fc1efab 100644
--- a/pycytominer/feature_select.py
+++ b/pycytominer/feature_select.py
@@ -43,10 +43,10 @@ def feature_select(
----------
profiles : pandas.core.frame.DataFrame or file
DataFrame or file of profiles.
- features : list
+ features : list, default "infer"
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
prefixed with "Cells", "Nuclei", or "Cytoplasm".
image_features: bool, default False
Whether the profiles contain image features.
diff --git a/pycytominer/normalize.py b/pycytominer/normalize.py
index 7a83ca5f..06c55fb6 100644
--- a/pycytominer/normalize.py
+++ b/pycytominer/normalize.py
@@ -34,14 +34,15 @@ def normalize(
features : list
A list of strings corresponding to feature measurement column names in the
`profiles` DataFrame. All features listed must be found in `profiles`.
- Defaults to "infer". If "infer", then assume cell painting features are those
+ Defaults to "infer". If "infer", then assume features are from CellProfiler output and
prefixed with "Cells", "Nuclei", or "Cytoplasm".
image_features: bool, default False
Whether the profiles contain image features.
meta_features : list
A list of strings corresponding to metadata column names in the `profiles`
DataFrame. All features listed must be found in `profiles`. Defaults to "infer".
- If "infer", then assume metadata features are those prefixed with "Metadata"
+ If "infer", then assume CellProfiler metadata features, identified by
+ column names that begin with the `Metadata_` prefix."
samples : str
The metadata column values to use as a normalization reference. We often use
control samples. The function uses a pd.query() function, so you should
@@ -114,7 +115,7 @@ def normalize(
normalized_df = normalize(
profiles=data_df,
features=["x", "y", "z", "zz"],
- meta_features="infer",
+ meta_features=["Metadata_plate", "Metadata_treatment"],
samples="Metadata_treatment == 'control'",
method="standardize"
)
diff --git a/pycytominer/operations/correlation_threshold.py b/pycytominer/operations/correlation_threshold.py
index 7c4522ba..a888a012 100644
--- a/pycytominer/operations/correlation_threshold.py
+++ b/pycytominer/operations/correlation_threshold.py
@@ -20,9 +20,10 @@ def correlation_threshold(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/operations/get_na_columns.py b/pycytominer/operations/get_na_columns.py
index ad36c377..f288f2cd 100644
--- a/pycytominer/operations/get_na_columns.py
+++ b/pycytominer/operations/get_na_columns.py
@@ -14,9 +14,10 @@ def get_na_columns(population_df, features="infer", samples="all", cutoff=0.05):
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `profiles` DataFrame. All features listed must be found in `profiles`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
@@ -36,8 +37,8 @@ def get_na_columns(population_df, features="infer", samples="all", cutoff=0.05):
if features == "infer":
features = infer_cp_features(population_df)
- else:
- population_df = population_df.loc[:, features]
+
+ population_df = population_df.loc[:, features]
num_rows = population_df.shape[0]
na_prop_df = population_df.isna().sum() / num_rows
diff --git a/pycytominer/operations/noise_removal.py b/pycytominer/operations/noise_removal.py
index aba1a29e..e3e41923 100644
--- a/pycytominer/operations/noise_removal.py
+++ b/pycytominer/operations/noise_removal.py
@@ -22,9 +22,10 @@ def noise_removal(
The list of unique perturbations corresponding to the rows in population_df. For example,
perturb1_well1 and perturb1_well2 would both be "perturb1".
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pycytominer/operations/variance_threshold.py b/pycytominer/operations/variance_threshold.py
index 67d3b767..72da751d 100644
--- a/pycytominer/operations/variance_threshold.py
+++ b/pycytominer/operations/variance_threshold.py
@@ -18,9 +18,10 @@ def variance_threshold(
population_df : pandas.core.frame.DataFrame
DataFrame that includes metadata and observation features.
features : list, default "infer"
- List of features present in the population dataframe [default: "infer"]
- if "infer", then assume cell painting features are those that start with
- "Cells_", "Nuclei_", or "Cytoplasm_".
+ A list of strings corresponding to feature measurement column names in the
+ `population_df` DataFrame. All features listed must be found in `population_df`.
+ Defaults to "infer". If "infer", then assume CellProfiler features are those
+ prefixed with "Cells", "Nuclei", or "Cytoplasm".
samples : str, default "all"
List of samples to perform operation on. The function uses a pd.DataFrame.query()
function, so you should structure samples in this fashion. An example is
diff --git a/pyproject.toml b/pyproject.toml
index 4b5faa17..a00ee920 100644
--- a/pyproject.toml
+++ b/pyproject.toml
@@ -73,6 +73,13 @@ pytest-cov = "^4.1.0"
pre-commit = ">=3.3.2"
commitizen = "^3.12.0"
ruff = "^0.3.4"
+mypy = "^1.11.2"
+types-openpyxl = "^3.1.5.20240918"
+pandas-stubs = [
+ {version = "<2.2.2.240909", python = "<3.10"},
+ {version = "^2.2.2.240909", python = ">=3.10"}
+]
+types-sqlalchemy = "^1.4.53.38"
[tool.poetry.group.docs]
optional = true
@@ -177,6 +184,29 @@ preview = true
[tool.pytest.ini_options]
testpaths = "tests"
+[tool.mypy]
+exclude = [
+ # ignore notebook-based walkthroughs
+ "walkthroughs",
+ # ignore tests dir
+ "tests"
+]
+
+
+[[tool.mypy.overrides]]
+# ignore missing import errors for the following
+# packages which do not have standard type stubs
+# or experience issues with imported types.
+module = [
+ "sklearn.*",
+ "scipy.*",
+ "cytominer_database.*",
+ "fire.*",
+ "boto3.*",
+ "botocore.*"
+]
+ignore_missing_imports = true
+
[build-system]
requires = ["poetry-core>=1.7.0", "poetry-dynamic-versioning>=1.1.0"]
build-backend = "poetry_dynamic_versioning.backend"
diff --git a/tests/test_aggregate.py b/tests/test_aggregate.py
index d1a08889..7765ec31 100644
--- a/tests/test_aggregate.py
+++ b/tests/test_aggregate.py
@@ -64,6 +64,19 @@ def test_aggregate_median_allvar():
assert aggregate_result.equals(expected_result)
+ # Test that ImageNumber column would be dropped before return
+ data_df_with_imagenumber = data_df.copy()
+ data_df_with_imagenumber["ImageNumber"] = "1"
+
+ aggregate_result = aggregate(
+ population_df=data_df_with_imagenumber,
+ strata=["g"],
+ features="infer",
+ operation="median",
+ )
+
+ assert aggregate_result.equals(expected_result)
+
# Test output
aggregate(
population_df=data_df,
diff --git a/tests/test_cyto_utils/test_feature_infer.py b/tests/test_cyto_utils/test_feature_infer.py
index df839858..9c8e8e2a 100644
--- a/tests/test_cyto_utils/test_feature_infer.py
+++ b/tests/test_cyto_utils/test_feature_infer.py
@@ -39,10 +39,10 @@ def test_feature_infer():
def test_feature_infer_nocp():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
infer_cp_features(population_df=non_cp_data_df)
- assert "No CP features found." in str(nocp.value)
+ assert "No features or metadata found." in str(nocp.value)
def test_metadata_feature_infer():
diff --git a/tests/test_cyto_utils/test_output.py b/tests/test_cyto_utils/test_output.py
index 72d5a9cf..0aeefd06 100644
--- a/tests/test_cyto_utils/test_output.py
+++ b/tests/test_cyto_utils/test_output.py
@@ -56,6 +56,20 @@ def test_output_default():
result, DATA_DF, check_names=False, check_exact=False, atol=1e-3
)
+ # test with an output_type of None
+ output_result = output(
+ df=DATA_DF,
+ output_filename=output_filename,
+ compression_options=TEST_COMPRESSION_OPTIONS,
+ float_format=None,
+ output_type=None,
+ )
+ result = pd.read_csv(output_result)
+
+ pd.testing.assert_frame_equal(
+ result, DATA_DF, check_names=False, check_exact=False, atol=1e-3
+ )
+
def test_output_tsv():
# Test input filename of writing a tab separated file
diff --git a/tests/test_operations/test_correlation_threshold.py b/tests/test_operations/test_correlation_threshold.py
index 2ca393cc..9845b99f 100644
--- a/tests/test_operations/test_correlation_threshold.py
+++ b/tests/test_operations/test_correlation_threshold.py
@@ -75,7 +75,7 @@ def test_correlation_threshold_samples():
def test_correlation_threshold_featureinfer():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
correlation_threshold_result = correlation_threshold(
population_df=data_df,
features="infer",
@@ -84,7 +84,7 @@ def test_correlation_threshold_featureinfer():
method="pearson",
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
data_cp_df = data_df.copy()
data_cp_df.columns = [f"Cells_{x}" for x in data_df.columns]
diff --git a/tests/test_operations/test_get_na_columns.py b/tests/test_operations/test_get_na_columns.py
index 9c8bd557..48b5ab71 100644
--- a/tests/test_operations/test_get_na_columns.py
+++ b/tests/test_operations/test_get_na_columns.py
@@ -67,9 +67,9 @@ def test_get_na_columns_sample():
def test_get_na_columns_featureinfer():
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
get_na_columns(
population_df=data_df, samples="all", features="infer", cutoff=0.1
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
diff --git a/tests/test_operations/test_variance_threshold.py b/tests/test_operations/test_variance_threshold.py
index a1a19764..1d7cd481 100644
--- a/tests/test_operations/test_variance_threshold.py
+++ b/tests/test_operations/test_variance_threshold.py
@@ -102,12 +102,12 @@ def test_variance_threshold():
def test_variance_threshold_featureinfer():
unique_cut = 0.01
- with pytest.raises(AssertionError) as nocp:
+ with pytest.raises(ValueError) as nocp:
excluded_features = variance_threshold(
population_df=data_unique_test_df, features="infer", unique_cut=unique_cut
)
- assert "No CP features found." in str(nocp.value)
+ assert "No features found." in str(nocp.value)
data_cp_df = data_unique_test_df.copy()
data_cp_df.columns = [f"Cells_{x}" for x in data_unique_test_df.columns]