These materials provide a practical guide on analysing viral amplicon sequencing data for genomic surveillance, with a specific focus on SARS-CoV-2. While centered on SARS-CoV-2, the concepts and pipelines explored here are applicable to various viruses. The content includes the analysis of data from clinical isolates and wastewater samples. For clinical isolates, we illustrate how to create consensus sequences for upload to databases like GISAID and for downstream applications such as variant annotation and phylogeny. Wastewater sample analysis includes estimating variant and mutation frequencies. For both applications we will use a standardized bioinformatic pipeline compatible with both Illumina and Nanopore data. The materials cover assigning sequences to lineages, identifying variants of interest and creating visualizations to effectively communicate findings. Throughout, you will acquire foundational bioinformatic skills, including Unix command line usage and scripting for reproducible analyses.
+
+
+
+
+
+
+Learning Objectives
+
+
+
+
+
Recognise the uses of genomic surveillance to inform public health actions during a pandemic.
+
Assemble high-quality SARS-CoV-2 genome sequences starting with raw sequencing data from clinical isolates.
+
Assign consensus sequences to lineages and identify variants of interest/concern.
+
Capture high-quality metadata, recognising its impact on downstream analyses.
+
Construct phylogenetic trees to contextualise new samples in a set of background samples.
+
Estimate variant frequencies from mixed wastewater samples.
+
Produce visualisations to communicate your findings and help inform public health action.
+
+
+
+
+
Target Audience
+
These materials are aimed at life scientists and molecular lab technicians interested in the bioinformatic analysis of viral genomic data. In particular, it will benefit those working in SARS-CoV-2 sequencing facilities, such as public health labs.
+
+
+
Prerequisites
+
We assume no prior bioinformatics experience or experience with the tools introduced in this course. An elementary knowledge of molecular and viral biology is assumed (concepts such as: DNA, RNA, PCR, primers, SNPs).
+
+
+
Citation
+
+
Please cite these materials if:
+
+
You adapted or used any of them in your own teaching.
+
These materials were useful for your research work. For example, you can cite us in the methods section of your paper: “We carried our analyses based on the recommendations in Tavares et al (2022).”.
+
+
You can reference these materials as:
+
+
Tavares H, Salehe B, Kumar A, Castle M & UKHSA New Variant Assessment Platform Team (2022) “cambiotraining/sars-cov-2-genomics: Introduction to Sars-CoV-2 Genomics”, https://cambiotraining.github.io/sars-cov-2-genomics
+
+
Or, in BibTeX format:
+
@Misc{,
+ author = {Tavares, Hugo and Salehe, Bajuna and Kumar, Ankit and Castle, Matt and UKHSA New Variant Assessment Platform Team},
+ title = {cambiotraining/sars-cov-2-genomics: Introduction to Sars-CoV-2 Genomics},
+ month = {March},
+ year = {2022},
+ url = {https://cambiotraining.github.io/sars-cov-2-genomics},
+}
+
Please make sure to include a link to the materials in the citation. (we will add a DOI in due time)
+
The contributing members from University of Cambridge Bioinformatics Training Facility team are:
+
+
Matt Castle, Bioinformatics Training Manager
+
Hugo Tavares, Senior Teaching Associate
+
Bajuna Salehe, Teaching Associate
+
Ankit Kumar, Teaching Assistant
+
+
The UKHSA’s NVAP Team members that supported these materials are:
+
+
Dr Leena Inamdar, NVAP Programme Lead and Global Health Lead
+
Dr Babak Afrough, Senior Project Manager
+
Aude Wilhelm, Senior Epidemiology Scientist
+
Richard Myers, Data Analytics Surveillance Head Bioinformatician
Enumerate some examples of how the genomic surveillance of SARS-CoV-2 has impacted public health decisions during the ongoing pandemic.
+
Describe what a variant of concern (VOC) is and how it differs from a variant of interest (VOI).
+
Describe the sources of information that can be obtained from GISAID, Pango, Nextstrain and the WHO nomenclature systems.
+
Contrast different sequencing protocols (e.g. amplicon, metagenomic) and technologies (e.g. Illumina and Nanopore) commonly used for SARS-CoV-2 sequencing, including the pros and cons of each.
+
Understand the steps involved in the widespread ARTIC protocol and the differences between its versions.
+
List key metadata fields needed with each sample to make best use of the data and recognise some limitations related to privacy.
+
+
+
+
+
+
1.1 What is SARS-CoV-2?
+
SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) is a betacoronavirus resposible for the COVID-19 disease and caused a global outbreak leading to an ongoing pandemic. The initial spread of this virus started in the city of Wuhan, in China. Despite early efforts to contain its spread in China (through several lockdowns in the country), the virus spread to other provinces within China and, eventually, to other countries across the world. This led to the World Health Organisation (WHO) declaring a public health emergency on 30 January 2020 and then a pandemic on 11 March 2020.
+
SARS-CoV-2 is an RNA virus, composed of single-stranded RNA. The first SARS-CoV-2 genome was published in January 2020 and is approximately 30Kb long. It encodes several proteins including the so-called Spike protein (or ‘S’ for short), which is used by the virus to interact and eventually enter human cells and cause infection. This interaction happens by the binding of the S protein to the ACE2 protein receptor found in human cells.
The genome sequence of SARS-CoV-2 has been a huge contributor to our ability to manage current pandemic. Namely:
+
+
It allowed the development of a vaccine to target the S protein.
+
It allowed the development of diagnostic tests for positive cases (lateral flow test and PCR-based test).
+
It allowed the development of protocols for whole-genome sequencing of the virus from positive human samples.
+
+
This last point is the focus of our workshop, and we will spend some time looking at how to analyse these data.
+
You can watch the video below, created by the Maastrich University, if you want to learn more about the lifecycle of the SARS-CoV-2 virus and the role that its different proteins play in this context:
+
+
+
+
+
+
+
1.2 Genomic Surveillance
+
The number of SARS-CoV-2 genomes sequenced is the largest of a pathogen ever done. As such, it has enabled us to track how the virus evolves and spreads both at a local and global scale at an unprecendented resolution. This allowed the identification of mutations that affect characteristics of the virus, such as its transmissibility or severity of the disease, and testing new strains of the virus to understand whether they are effectively neutralised by current or future vaccines.
+
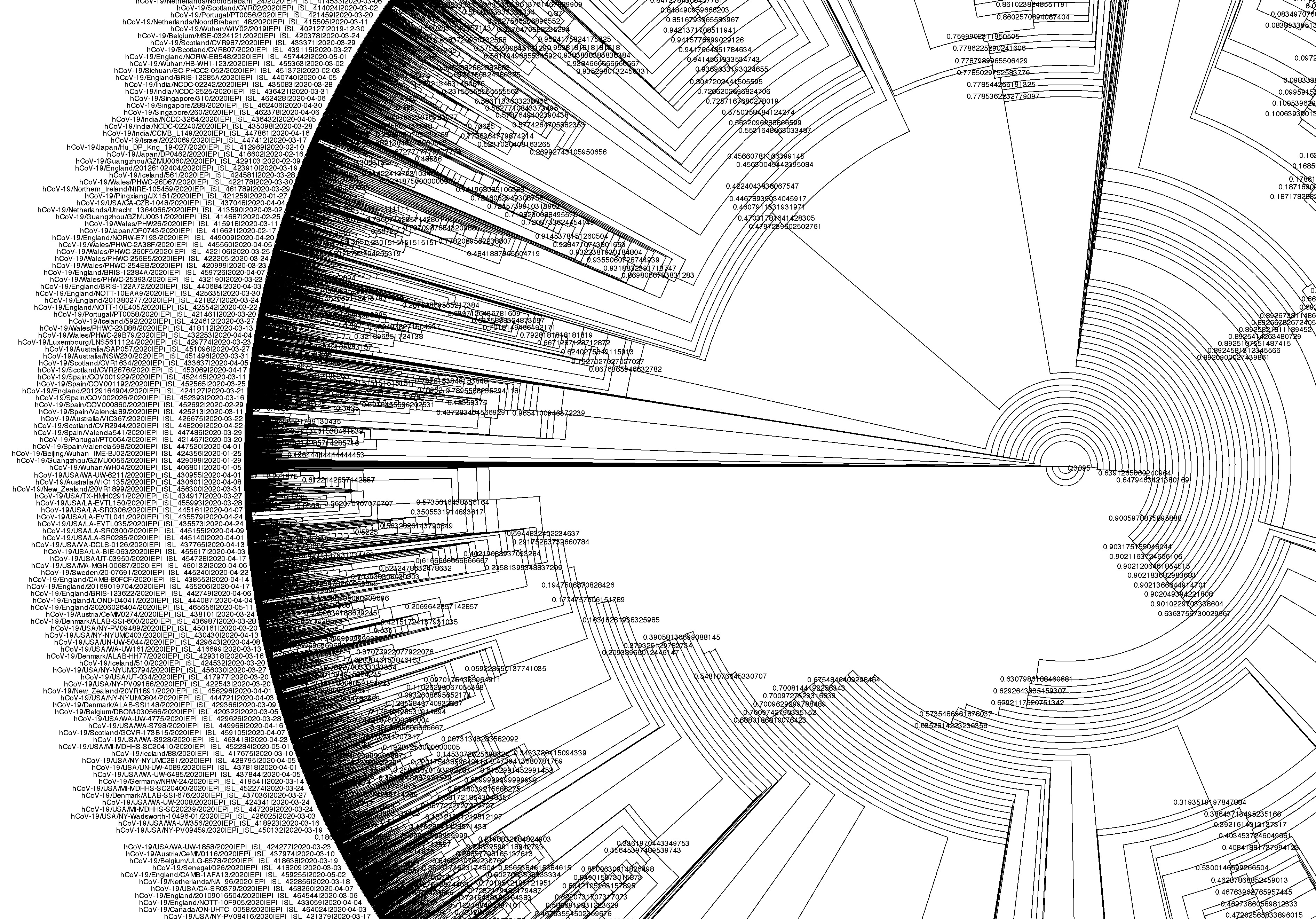
One of the main applications of SARS-CoV-2 sequencing is to infer relationships between the different circulating forms of the virus. This is done by comparing these sequences and building phylogenetic trees that reflect their sequence similarities. From these, it is possible to identify particular clusters of similar sequences that spread faster than expected, and may therefore be associated with mutations that increase the virus’ fitness.
+
+
+
+Example of global phylogeny from the Nextstrain public server. Colours show clusters of similar sequences. (Screenshot taken Feb 2022)
+
+
+
For example, one of the first mutations identified from these sequencing efforts was A23403G (an adenine was replaced by a guanine in position 23,403 of the genome), which caused an aminoacid substitution in the S protein that increased virus infectivity and transmissibility. Since then, many new forms of the virus have been identified as being of particular concern for public health, and have been classified by the WHO as Variants of Concern. These have been named based on letters of the Greek alphabet, and have been a crucial way to inform public health policy and containment measures around the world.
+
In addition to the WHO variant nomenclature, there are three main projects that have been instrumental in grouping SARS-CoV-2 sequences into similar groups:
+
+
The GISAID nomenclature classifies sequences based on key mutations that define particular groups of sequences in the global phylogeny. It also complements its classification by borrowing information from the Pango nomenclature system.
+
The Pango nomenclature is based on the phylogeny of SARS-CoV-2 and defined as groups of sequences that share a common ancestor and a distinctive sequence feature (for example, all share the same single nucleotide change). This nomenclature is also sometimes referred as “Rambaut et al. 2020” after the respective publication.
+
The Nextstrain nomenclature is slightly more informal than the above, its main purpose being to facilitate public health discussions. Despite this, it is still informed by the phylogenetic placement of sequences in a tree and therefore has a large overlap with the Pango nomenclature.
+
+
We will learn more about how sequences are classified into lineages and variants of concern in the section about Lineage Assignment.
+
+
+
+
+
+
+What is the difference: strain, lineage, clade, variant?
+
+
+
+
These terms are sometimes used interchangeably in informal conversations about the different forms of SARS-CoV-2. For our purposes, these are the definitions we will use:
+
A strain is a group of viruses that are sufficiently diverged from others, so that they are quite distinct at a sequence level as well as in their biological properties. SARS-CoV-2 is still considered to be a single strain. Examples of other coronavirus strains are SARS-CoV and MERS-CoV.
+
The terms lineage and clade are somewhat similar, in that both represent groups of similar sequences, inferred from a phylogenetic analysis and sharing a common ancestor. Their main difference (at least in the current SARS-CoV-2 nomenclature systems) is the level of resolution. Clades tend to be more broadly defined and therefore include more sequences within it. They are useful to discuss long-term trends during a pandemic. Lineages have a finer resolution, being useful to identify patterns related to a recent outbreak.
+
Since a phylogenetic tree is inherently hierarchical, there isn’t always a clear-cut way of defining where one lineage/clade starts and another ends. That is why lineage classification is a partially manual process, involving human curation by experts.
+
Finally, the term variant is usually used to refer to WHO’s variants of interest or variants of concern (e.g. the Alpha, Delta and Omicron variants). Variants are distinct from each other by the combination of all sequence changes in their genomes. The term “variant” can be ambiguous when used in the field of bioinformatics, and we return to this in the section about Lineage Assigment
+
+
+
+
+
1.3 The GISAID Database
+
The widespread use of genome sequences would have not been possible without the ability to centrally collect these sequences and make them available to researchers and public health professionals. The main repository used to deposit SARS-CoV-2 sequences is the database managed by the GISAID Initiative. This allows sharing the sequencing data as well as metadata associated with it (such as dates of collection, geo-location, patient information, etc.). For example, the outbreak.info website that we just used in the exercise above directly pulls data from GISAID to update its reports on a daily basis.
+
At the end of this course you too will be able to contribute to this database, by producing full genome sequences assembled from sequencing data.
+
+
+
+
+
+
+Create a GISAID account
+
+
+
+
Go to the GISAID registration page and create an account, so you can gain access to the data stored in GISAID as well as the ability to submit your own sequences in the future.
+
+
+
+
+
1.4 SARS-CoV-2 Sequencing
+
Routine SARS-CoV-2 sequencing is done with a method generally referred to as amplicon sequencing. This method relies on amplifying the genetic material using polymerase chain reaction (PCR) with a panel of primers designed against the known SARS-CoV-2 genome.
+
One of the most popular methods for preparing virus RNA for sequencing is amplicon sequencing. This method consists of amplifying the genetic material by PCR using a panel of primers that covers the entire genome of the target virus. This is required, since the starting amount of viral genetic material in a sample is typically very low (most of the material will belong to the host - in this case, human RNA).
+
The most popular protocol for amplicon sequencing has been developed by the ARTIC network, whose aim is to develop standardised protocols for viral sample processing. The group has designed and tested a panel of primers that work well to amplify the SARS-CoV-2 genome in a mostly unbiased way. This is a challenging task, as the protocol involves pooling hundreds of primers together in a single reaction! Also, as the virus mutates in the population, primers that used to work in the original template genome may no longer work in the new variants circulating in the population (“primer erosion”). Therefore, the ARTIC primers have gone through several versions, which are updated and optimised to work on the most common circulating lineages. These are called “primer schemes” and are made publicly available in a public repository.
+
+
+
+Schematic of the ARTIC protocol. The RNA is reverse-transcribed to cDNA, then two pools of primers (that cover the entire SARS-CoV-2 genome) are used to PCR-amplify the material. This material is used to prepare sequencing libraries for the relevant platform that will be used downstream (Illumina or Nanopore). Source: Gohl et al. 2020
+
+
+
Besides amplicon sequencing, other methods can also be used to obtain SARS-Cov-2 genomes:
+
+
Metagenomic RNA sequencing was the method used to assemble the first SARS-CoV-2 genome and one of the first sequences in Cambodia. This method consists of extracting viral RNA (using commercially available kits) followed by high-throughput RNA-seq. The resulting sequences are then compared with nucleotide databases of known organisms, and viral sequences selected for de-novo assembly. This approach is suitable when the virus sequence is unknown, but requires a high sequencing depth, increasing its costs.
+
Sequence capture protocols are also available, whereby the samples are enriched for the target virus by using a panel of probes against the SARS-CoV-2 genome, followed by sequencing and reference-based assembly. This approach is more similar to amplicon sequencing, as it works by enriching the sample for the known virus.
+
+
+
+
+Simplified diagram of the main steps involved in metagenomic and amplicon sequencing approaches. In amplicon sequencing the cDNA material is first PCR-amplified with virus-specific primers to enrich the sample, followed by sequencing and downstream bioinformatic analysis. With the metagenomic approach the mixed cDNA material is sequenced and the resulting sequences are bioinformatically separated between known sequences (from other organisms) and unknown sequences. The unknown sequences can then be de-novo assembled into a new genome.
+
+
+
Despite these alternative methods, amplicon sequencing remains one of the most popular methods for large-scale viral surveillance due to its low cost and high-throughput. The data generated from this method will be the focus of this course.
+
+
+
+
+
+
+Illumina or Nanopore?
+
+
+
+
Both of these sequencing platforms can be used to sequence amplicon samples.
+
Nanopore platforms allow sequencing 96 samples at a time, are readily available as portable devices, and have fast run times. This gives them great flexibility, making them an excellent solution for rapidly building sequencing capacity in a lab.
+
By comparison, Illumina platforms give higher throughput, are cheaper to run per sample and have lower error rates. However, they require substantial upfront cost to setup and equip in the lab and take longer to run.
+
+
+
+
+
+
+
+
+Alternative Amplicon Sequencing Protocols
+
+
+
+
Although the ARTIC protocol is one of the most popular used in routine SARS-CoV-2 sequencing, there are alternative sets of primers and protocols available.
+
For example, Thermo Fisher has the Ion AmpliSeq SARS-CoV-2 kit, designed to work with Ion Torrent sequencing platforms.
+
An alternative protocol that has been developed by the community is the midnight protocol. This protocol consists of amplifying larger PCR fragments, thus requiring fewer primer pairs than the ARTIC protocol. This leads to a lower complexity in the multiplex PCR reaction and fewer chances of PCR dropout due to mis-priming againts new variants. However, because it uses longer PCR fragments, it can only be used with long-read sequencing (Nanopore) and not short-read sequencing (Illumina).
There are two big considerations when collecting samples for sequencing:
+
+
Is there enough viral material in the sample (viral load)?
+
Did I collect all the necessary information about each sample (metadata)?
+
+
+
1.5.1 Viral Load
+
When collecting patient samples for sequencing, it is important to quantify the viral load in the sample. This is usually done by quantitative RT-PCR (RT-qPCR), whereby the amplification of the samples is monitored by measuring a dye that is incorporated during the PCR reaction. This results in amplification curves, which reflect the amount of RNA present in the sample. The more PCR cycles are needed to saturate the signal, the lower the amount of virus in the sample.
+
+
+
+Overview of RT-qPCR quantification of viral load in a sample. Adapted from Smyrlaki et al. 2020
+
+
+
The result of RT-qPCR is usually expressed as the PCR cycle at which a particular threshold in the amplification curve is reached, the Ct value. Generally, samples with Ct > 30 are not worth sequencing, as their genome completeness is likely going to be low due to the low amounts of starting material.
One important consideration when collecting samples, is to record as much information as possible about each sample. The Public Health Alliance for Genomic Epidemiology (PHA4GE) coalition provides several guidelines and a protocol to aid in metadata collection. There are also essential metadata needed to upload new SARS-CoV-2 genome sequences to the GISAID database (Figure).
+
+
+
+Snapshot of the GISAID upload template metadata sheet.
+
+
+
Two key pieces of information for genomic surveillance are the date of sample collection and the geographic location of that sample. This information can be used to understand which variants are circulating in an area at any given time.
+
Privacy concerns need to be considered when collecting and storing sensitive data. However, it should be noted that sensitive data can still be collected, even if it is not shared publicly (e.g. via the GISAID database). Such sensitive information may still be useful for the relevant public health authorities, who may use those sensitive information for a finer analysis of the data. For example, epidemiological analysis will require individual-level metadata (“person, place, time”) to be available, in order to track the dynamics of transmission within a community.
+
This is the general advice when it comes to metadata collection: record as much information about each sample as possible!
+
+
+
+
1.6 SARS-CoV-2 Bioinformatics
+
What bioinformatic skills do we need in order to analyse SARS-CoV-2 genome sequencing data? While there are several software tools that have been specifically developed for SARS-CoV-2 analysis (and we will see some of them in this course), there is a set of foundational skills that are applicable to any bioinformatics application:
+
+
The use of the Unix command line. Linux is the most common operating system for computational work and most of the bioinformatic software only runs on it.
+
Getting familiar with common file formats in bioinformatics. This includes files to store nucleotide sequences, sequence alignments to a reference genome, gene annotations, phylogenetic trees, amongst others.
+
Understand software tools’ documentation and how to configure different options when running our analyses.
+
+
We will turn to these topics in the following sessions.
+
+
+
1.7 Exercises
+
+
+
+
+
+
+WHO Variants
+
+
+
+
+
+
+
+
+
Looking through the WHO variants page, can you find what the difference is between a Variant of Interest (VOI) and a Variant of Concern (VOC)?
+
Can you find the correspondence between the VOCs and their respective lineages in other classification systems (GISAID, Nextstrain and Pango)?
+
Go to the outbreak.info website and search for a country of your choice (for example, your country of origin or where you live).
+
How many sequences are available from the last 360 days? (Note: by default only the last 60 days are shown. You can change this in the text box found on the right of the report page.)
+
What were the most common lineages of the virus in circulation in the last 360 days? Do you notice sharp changes in the frequency of WHO Variants of Concern?
+
+
+
+
+
+
+
+
+
+
+
+
+Metadata
+
+
+
+
+
+
+
+
Open the folder under “Course_Materials > 01-Unix > metadata”, where you will find several CSV files with information about samples that were sequenced and will be analysed by us later in the course. Double-click to open one of these files (any of them is fine), which should open them on a spreadsheet software (on our training machines it is LibreOffice, but Excel would also work).
+
The column names of these files are based on the PHA4GE nomenclature system.
From that link, go to the Supporting Materials to find the “field mappings” spreadsheet, which matches naming conventions between PHA4GE and other databases such as GISAID.
+
Based on this information, can you match the column names from our sample information table to the fields required by GISAID?
+
What do you think is the naming convention for the isolate name? How would you adjust this when uploading the data to GISAID?
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
The first step of this protocol provides with a link to a GitHub repository containing several template files: https://github.com/pha4ge/SARS-CoV-2-Contextual-Data-Specification.
+
If we follow that link, we will see several spreadsheet files (for Excel or LibreOffice) with templates that can be used to record samples metadata. In the “Supporting Materials” we find a link to “PHA4GE to WHO and sequence repository field mappings”. If we download and open that file we will see a spreadhseet with 4 tabs. Each of the tabs shows the equivalence between the PHA4GE column names and the equivalent names used by other platforms.
+
Looking at the equivalence between the column names in our metadata sheets and the GISAID nomenclature, we can identify only some of them:
+
+
+
+
+
+
+
+
+
Our Name
+
GISAID equivalent
+
Comments
+
+
+
+
+
sample
+
+
+
+
+
sample_collection_date
+
Collection date
+
+
+
+
geo_loc_country
+
Location
+
+
+
+
geo_loc_region
+
Location
+
In GISAID the field “Location” is used for “Continent / Country or Territory / Region”
+
+
+
latitude
+
+
+
+
+
longitude
+
+
+
+
+
sequencing_instrument
+
Sequencing technology
+
+
+
+
sample_collection_year
+
+
+
+
+
sample_collection_month
+
+
+
+
+
sample_collection_day
+
+
+
+
+
organism
+
+
+
+
+
isolate
+
Virus name
+
+
+
+
sequencing_protocol_name
+
+
+
+
+
amplicon_pcr_primer_scheme
+
+
+
+
+
sequencing_protocol
+
+
+
+
+
+
Looking at this spreadhseet, we can also see this explanation about the “Virus Name” field:
+
+
While the meanings and structures of the GISAID field “Virus name” and the PHA4GE field “isolate” overlap, GISAID requires a slightly different structure for Virus name. e.g. PHA4GE structure: SARS-CoV-2/country/sampleID/2020, GISAID structure: hCov-19/country/sampleID/2020. Change “SARS-CoV-2” to “hCov-19” in the isolate name.
+
+
So, in order to upload our data to GISAID we would need to change the name of the virus accordingly.
+
+
+
+
+
+
+
+
+
+
+
+
+
1.8 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
The sequencing of SARS-CoV-2 genomes has allowed the tracking of new variants throughout the pandemic.
+
The World Health Organisation defines Variants of Concern as SARS-CoV-2 forms with characteristics of public health concern. This includes increased transmissibility, virulence, disease symptoms or vaccine resistance.
+
GISAID, Pango and Nextstrain are organisations whose work includes the classification of SARS-CoV-2 genomes into groups that may later be classified as variants of concern by WHO.
+
GISAID also plays a key role as the main database for assembled SARS-CoV-2 genomes, submitted by the community.
+
Routine SARS-CoV-2 sequencing is usually done by amplicon sequencing (amplifying an infected sample by PCR using primers that cover the entire genome). The most popular protocol has been developed by the ARTIC network.
+
Other methods of sequencing include metagenomic and sequence capture. However, these methods require more sequencing and therefore are not commonly used for sequencing at a population scale.
+
Both Illumina and Nanopore platforms can be used for sequencing SARS-CoV-2 amplicon samples.
+
Metadata collection is essential for interpreting the results of the sequencing.
+
+
For genomic surveillance purposes recording of geographic location and date of sampling are crucial.
+
Other useful information includes details about the sequencing (e.g. sample preparation protocols and sequencing platforms used).
Describe differences between sequencing data produced by Illumina and Nanopore platforms.
+
Recognise the structure of common file formats in bioinformatics, in particular FASTA and FASTQ files.
+
Use FastQC to produce a quality report for Illumina sequences.
+
Use MultiQC to produce a report compiling multiple quality statistics.
+
Examine quality reports to identify problematic samples.
+
+
+
+
+
+
2.1 Next Generation Sequencing
+
The sequencing of genomes has become more routine due to the rapid drop in DNA sequencing costs seen since the development of Next Generation Sequencing (NGS) technologies in 2007. One main feature of these technologies is that they are high-throughput, allowing one to more fully characterise the genetic material in a sample of interest.
+
There are three main technologies in use nowadays, often referred to as 2nd and 3rd generation sequencing:
+
+
Illumina’s sequencing by synthesis (2nd generation)
The video below from the iBiology team gives a great overview of these technologies.
+
+
+
+
+
2.1.1 Illumina Sequencing
+
Illumina’s technology has become a widely popular method, with many applications to study transcriptomes (RNA-seq), epigenomes (ATAC-seq, BS-seq), DNA-protein interactions (ChIP-seq), chromatin conformation (Hi-C/3C-Seq), population and quantitative genetics (variant detection, GWAS), de-novo genome assembly, amongst many others.
+
An overview of the sequencing procedure is shown in the animation video below. Generally, samples are processed to generate so-called sequencing libraries, where the genetic material (DNA or RNA) is processed to generate fragments of DNA with attached oligo adapters necessary for the sequencing procedure (if the starting material is RNA, it can be converted to DNA by a step of reverse transcription). Each of these DNA molecule is then sequenced from both ends, generating pairs of sequences from each molecule, i.e. paired-end sequencing (single-end sequencing, where the molecule is only sequenced from one end is also possible, although much less common nowadays).
+
This technology is a type of short-read sequencing, because we only obtain short sequences from the original DNA molecules. Typical protocols will generate 2x50bp to 2x250bp sequences (the 2x denotes that we sequence from each end of the molecule).
+
+
+
+
The main advantage of Illumina sequencing is that it produces very high-quality sequence reads (current protocols generate reads with an error rate of less than <1%) at a low cost. However, the fact that we only get relatively short sequences means that there are limitations when it comes to resolving particular problems such as long sequence repeats (e.g. around centromeres or transposon-rich areas of the genome), distinguishing gene isoforms (in RNA-seq), or resolving haplotypes (combinations of variants in each copy of an individual’s diploid genome).
+
+
+
2.1.2 Nanopore Sequencing
+
Nanopore sequencing is a type of long-read sequencing technology. The main advantage of this technology is that it can sequence very long DNA molecules (up to megabase-sized), thus overcoming the main shortcoming of short-read sequencing mentioned above. Another big advantage of this technology is its portability, with some of its devices designed to work via USB plugged to a standard laptop. This makes it an ideal technology to use in situations where it is not possible to equip a dedicated sequencing facility/laboratory (for example, when doing field work).
+
+
+
+Overview of Nanopore sequencing showing the highly-portable MinION device. The device contains thousands of nanopores embeded in a membrane where current is applied. As individual DNA molecules pass through these nanopores they cause changes in this current, which is detected by sensors and read by a dedicated computer program. Each DNA base causes different changes in the current, allowing the software to convert this signal into base calls.
+
+
+
One of the bigger challenges in effectively using this technology is to produce sequencing libraries that contain high molecular weight, intact, DNA. Another disadvantage is that, compared to Illumina sequencing, the error rates at higher, at around 5%.
+
+
+
+
+
+
+Illumina or Nanopore for SARS-CoV-2 sequencing?
+
+
+
+
Both of these platforms have been widely popular for SARS-CoV-2 sequencing. They can both generate data with high-enough quality for the assembly and analysis of SARS-CoV-2 genomes. Mostly, which one you use will depend on what sequencing facilities you have access to.
+
While Illumina provides the cheapest option per sample of the two, it has a higher setup cost, requiring access to the expensive sequencing machines. On the other hand, Nanopore is a very flexible platform, especially its portable MinION devices. They require less up-front cost allowing getting started with sequencing very quickly in a standard molecular biology lab.
+
+
+
+
+
+
2.2 Sequencing Analysis
+
In this section we will demonstrate two common tasks in sequencing data analysis: sequence quality control and mapping to a reference genome. There are many other tasks involved in analysing sequencing data, but looking at these two examples will demonstrate the principles of running bioinformatic programs. We will later see how bioinformaticians can automate more complex analyses in the consensus assembly section.
+
One of the main features in bioinformatic analysis is the use of standard file formats. It allows software developers to create tools that work well with each other. For example, the raw data from Illumina and Nanopore platforms is very different: Illumina generates images; Nanopore generates electrical current signal. However, both platforms come with software that converts those raw data to a standard text-based format called FASTQ.
+
+
2.2.1 FASTQ Files
+
FASTQ files are used to store nucleotide sequences along with a quality score for each nucleotide of the sequence. These files are the typical format obtained from NGS sequencing platforms such as Illumina and Nanopore (after basecalling).
In FASTQ files each sequence is always represented across 4 lines. The quality scores are encoded in a compact form, using a single character. They represent a score that can vary between 0 and 40 (see Illumina’s Quality Score Encoding). The reason single characters are used to encode the quality scores is that it saves space when storing these large files. Software that work on FASTQ files automatically convert these characters into their score, so we don’t have to worry about doing this conversion ourselves.
+
The quality value in common use is called a Phred score and it represents the probability that the respective base is an error. For example, a base with quality 20 has a probability \(10^{-2} = 0.01 = 1\%\) of being an error. A base with quality 30 has \(10^{-3} = 0.001 = 0.1\%\) chance of being an error. Typically, a Phred score threshold of >20 or >30 is used when applying quality filters to sequencing reads.
+
Because FASTQ files tend to be quite large, they are often compressed to save space. The most common compression format is called gzip and uses the extension .gz. To look at a gzip file, we can use the command zcat, which decompresses the file and prints the output as text.
+
For example, we can use the following command to count the number of lines in a compressed FASTQ file:
+
zcat sequences.fq.gz |wc-l
+
If we want to know how many sequences there are in the file, we can divide the result by 4 (since each sequence is always represented across four lines).
+
+
+
2.2.2 FASTQ Quality Control
+
One of the most basic tasks in Illumina sequence analysis is to run a quality control step on the FASTQ files we obtained from the sequencing machine.
+
The program used to assess FASTQ quality is called FastQC. It produces several statistics and graphs for each file in a nice report that can be used to identify any quality issues with our sequences.
For example, looking at the “Per base sequence quality” section for one of our samples, we can see a very high quality score, which is typical of Illumina data nowadays.
+
+
+
+Sequence quality plot from FastQC for one of our samples. The blue line shows the average across all samples. This sample is very high quality as all sequences have quality > 20 across the entire length of the reads.
+
+
+
+
+
+
+
+
+Quality Control Nanopore Reads
+
+
+
+
Although FastQC can run its analysis on any FASTQ files, it has mostly been designed for Illumina data. You can still run FastQC on basecalled Nanopore data, but some of the output modules may not be as informative. FastQC can also run on FAST5 files, using the option --nano.
+
You can also use MinIONQC, which takes as input the sequence_summary.txt file, which is a standard output file from the Guppy software used to convert Nanopore electrical signal to sequence calls.
+
+
+
+
+
2.2.3 Read Mapping
+
A common task in processing sequencing reads is to align them to a reference genome, which is typically referred to as read mapping or read alignment. We will continue exemplifying how this works for Illumina data, however the principle is similar for Nanopore data (although the software used is often different, due to the higher error rates and longer reads typical of these platforms).
+
Generally, these are the steps involved in read mapping (figure below):
+
+
Genome Indexing | Because reference genomes can be quite long, most mapping algorithms require that the genome is pre-processed, which is called genome indexing. You can think of a genome index in a similar way to an index at the end of a textbook, which tells you in which pages of the book you can find certain keywords. Similarly, a genome index is used by mapping algorithms to quickly search through its sequence and find a good match with the reads it is trying to align against it. Each mapping software requires its own index, but we only have to generate the genome index once.
+
Read mapping | This is the actual step of aligning the reads to a reference genome. There are different popular read mapping programs such as bowtie2 or bwa. The input to these programs includes the genome index (from the previous step) and the FASTQ file(s) with reads. The output is an alignment in a file format called SAM (text-based format - takes a lot of space) or BAM (compressed binary format - much smaller file size).
+
BAM Sorting | The mapping programs output the sequencing reads in a random order (the order in which they were processed). But, for downstream analysis, it is good to sort the reads by their position in the genome, which makes it faster to process the file.
+
BAM Indexing | This is similar to the genome indexing we mentioned above, but this time creating an index for the alignment file. This index is often required for downstream analysis and for visualising the alignment with programs such as IGV.
+
+
+
+
+Diagram illustrating the steps involved in mapping sequencing reads to a reference genome. Mapping programs allow some differences between the reads and the reference genome (red mutation shown as an example). Before doing the mapping, reads are usually filtered for high-quality and to remove any sequencing adapters. The reference genome is also indexed before running the mapping step. The mapped file (BAM format) can be used in many downstream analyses. See text for more details.
+
+
+
We have already prepared the SARS-CoV-2 genome index for the bowtie2 aligner. We have also prepared a shell script with the code to run the three steps above as an example. Let’s look at the content of this file (you can open it with nano scripts/mapping.sh):
In the first step, mapping, we are using two tools: bowtie2 and samtools. bowtie2 is the mapping program and samtools is a program used to manipulate SAM/BAM alignment files. In this case we used the | pipe to send the output of bowtie2 directly to samtools:
+
+
-x is the prefix of the reference genome index.
+
-1 is the path to the first read in paired-end sequencing.
+
-2 is the path to the second read in paired-end sequencing.
+
--threads 5 indicates we want to use 5 CPUs (or threads) to do parallel processing of the data.
+
| is the pipe that sends the output from bowtie2 to samtools sort.
+
-o is the name of the output file. By setting the file extension of this file to .bam, samtools will automatically save the file in the compressed format (which saves a lot of space).
+
The - symbol at the end of the samtools command indicates that the input is coming from the | pipe.
+
+
We also have a step that creates an index file for the BAM file using samtools index. This creates a file with the same name and .bai extension.
+
Finally, the script also contains a step that collects some basic statistics from the alignment, which we save in a text file. We will see how this file can be used to produce a quality control report below.
+
+
+
2.2.4 Visualising BAM Files in IGV
+
One thing that can be useful is to visualise the alignments produced in this way. We can use the program IGV (Integrative Genome Viewer) to do this:
+
+
Open IGV and go to File → Load from file….
+
In the file browser that opens go to the folder results/bowtie2/ and select the file ERR6129126.bam to open it.
+
Go back to File → Load from file… and this time load the BED files containing the primer locations. These can be found in resources/primers/artic_primers_pool1.bed and resources/primers/artic_primers_pool2.bed.
+
+
There are several ways to search and browse through our alignments, exemplified in the figure below.
+
+
+
+Screenshot IGV program. The search box at the top can be used to go to a specific region in the format “CHROM:START-END”. In the case of SARS-CoV-2 there is only one “chromosome” called NC_045512.2 (this is the name of the reference genome), so if we wanted to visualise the region between positions 21563 and 25384 (the Spike gene) we would write “NC_045512.2:21563-25384”.
+
+
+
+
+
2.2.5 Quality Reports
+
We’ve seen the example of using the program FastQC to assess the quality of our FASTQ sequencing files. And we have also seen an example of using the program samtools stats to obtain some quality statistics of our read mapping step.
+
When processing multiple samples at once, it can become hard to check all of these quality metrics individually for each sample. This is the problem that the software MultiQC tries to solve. This software automatically scans a directory and looks for files it recognises as containing quality statistics. It then compiles all those statistics in a single report, so that we can more easily look across dozens or even hundreds of samples at once.
+
Here is the command to run MultiQC and compile several quality statistics into a single report:
MultiQC generates a report, in this example in results/multiqc/multiqc_report.html. From this report we can get an overview of the quality across all our samples.
+
+
+
+Snapshot of some of the report sections from MultiQC. In this example we can see the “General Statistics” table and the “Sequence Quality Histograms” plot. One of the samples has lower quality towards the end of the read compared to other samples (red line in the bottom panel).
+
+
+
For example, from the section “General Statistics” we can see that the number of reads varies a lot between samples. Sample ERR5926784 has around 0.1 million reads, which is substantially lower than other samples that have over 1 million reads. This may affect the quality of the consensus assembly that we will do afterwards.
+
From the section “Sequence Quality Histograms”, we can see that one sample in particular - ERR5926784 - has lower quality in the second pair of the read. We can open the original FastQC report and confirm that several sequences even drop below a quality score of 20 (1% change of error). A drop in sequencing quality towards the end of a read can often happen, especially for longer reads. Usually, analysis workflows include a step to remove reads with low quality so these should not affect downstream analysis too badly. However, it’s always good to make a note of potentially problematic samples, and see if they produce lower quality results downstream.
+
+
+
+
2.3 Bioinformatic File Formats
+
Like we said above, bioinformatics uses many standard file formats to store different types of data. We have just seen two of these file formats: FASTQ for sequencing reads and BAM files to store reads mapped to a genome.
+
Another very common file format is the FASTA file, which is the format that our reference genome is stored as. The consensus sequences that we will generate are also stored as FASTA files. We detail this format below, but there are many other formats. Check out our appendix page on File Formats to learn more about them.
+
+
2.3.1 FASTA Files
+
Another very common file that we should consider is the FASTA format. FASTA files are used to store nucleotide or amino acid sequences.
+
The general structure of a FASTA file is illustrated below:
+
>sample01 <-- NAME OF THE SEQUENCE
+AGCGTGTACTGTGCATGTCGATG <-- SEQUENCE ITSELF
+
Each sequence is represented by a name, which always starts with the character >, followed by the actual sequence.
+
A FASTA file can contain several sequences, for example:
Each sequence can sometimes span multiple lines, and separate sequences can always be identified by the > character. For example, this contains the same sequences as above:
+
>sample01 <-- FIRST SEQUENCE STARTS HERE
+AGCGTGTACTGT
+GCATGTCGATG
+>sample02 <-- SECOND SEQUENCE STARTS HERE
+AGCGTGTACTGT
+GCATGTCGATG
+
To count how many sequences there are in a FASTA file, we can use the following command:
+
grep">" sequences.fa |wc-l
+
In two steps:
+
+
find the lines containing the character “>”, and then
+
count the number of lines of the result.
+
+
We will see FASTA files several times throughout this course, so it’s important to be familiar with them.
+
+
+
+
2.4 Exercises
+
+
+
+
+
+
+Sequence quality control: FASTQC
+
+
+
+
+
+
+
+
In the course materials directory 02-ngs/ we have several FASTQ files that we will use to assemble SARS-CoV-2 genomes. But first, we will run FastQC to check the quality of these files.
+
This is the basic command we could use in our samples:
Create the output directory for the analysis (results/fastqc).
+
+
+Hint
+
+The command to create directories is mkdir. By default, the mkdir directory only creates one directory at a time. In this case we need to create first the results directory and then the results/fastqc within it. Alternatively, both directories can be created at once using the -p option.
+
+
Modify the fastqc command shown above to add an option to run the analysis using 8 threads in parallel (or CPUs). Check the tool’s help (fastqc --help) to see what the option to do this is called.
+
Run the command. You will know it is running successfully because it prints progress of its analysis on the screen.
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
First, we can create a directory to output our results:
+
mkdir-p results/fastqc
+
The -p option ensures that both directories are created in one step. Otherwise, since the parent directory results did not exist, mkdir would throw an error.
+
To check the options available with FastQC we can run fastqc --help to get the complete documentation. As we scroll through the options, we can see the relevant one for running the analysis in parallel:
+
-t --threads Specifies the number of files which can be processed
+ simultaneously. Each thread will be allocated 250MB of
+ memory so you shouldn't run more threads than your
+ available memory will cope with, and not more than
+ 6 threads on a 32 bit machine
+
Although the documentation is a little technical, this means that if we have multiple CPUs available on our computer, we can set this option to allow multiple files to be processed in parallel. Our training machines have 8 CPUs, so we can run the command as follows:
The analysis report generated by FastQC is given as a .html file (opens in a web browser). We will go through the details of this below.
+
+
+
+
+
+
+
+
+
+
+
+
+
2.5 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
Illumina sequencing produces short reads (50bp - 200bp), typically from both ends of a DNA fragment. It is a comparatively cheap sequencing platform which produces very high-quality sequences.
+
Nanopore sequencing produces very long reads (typically hundreds of kilobases long). It is comparatively more expensive and has higher error rates. However, it is more flexible with some of its platforms being fully portable.
+
Sequencing reads are stored in a file format called FASTQ. This file contains both the nucleotide sequence and quality of each base.
+
The quality of Illumina sequence reads can be assessed using the software FastQC.
+
One common task in bioinformatics is to align or map reads to a reference genome. This involves:
+
+
Creating a genome index - this only needs to be done once.
+
Mapping the reads to the reference genome (e.g. using bowtie2) - the output is in SAM format.
+
Sorting the reads in the mapped file (using samtools sort) - the output is in BAM format.
+
Indexing the BAM alignment file (using samtools index).
+
+
The software MultiQC can be used to generate a single reports that compiles statistics across several samples.
+
Bioinformatics uses many standard file formats. One of the most common ones is the FASTA format, which is used to store nucleotide or amino acid sequences (no quality information is contained in these files). This is a standard format that assembled genomes are stored as.
+
+
+
+
+
\ No newline at end of file
diff --git a/materials/01-intro/images/Ct_coverage_relationship.png b/materials/01-intro/images/Ct_coverage_relationship.png
new file mode 100644
index 0000000..b70d12b
Binary files /dev/null and b/materials/01-intro/images/Ct_coverage_relationship.png differ
diff --git a/materials/01-intro/images/artic_protocol.png b/materials/01-intro/images/artic_protocol.png
new file mode 100644
index 0000000..5250977
Binary files /dev/null and b/materials/01-intro/images/artic_protocol.png differ
diff --git a/materials/01-intro/images/fastqc_quality.png b/materials/01-intro/images/fastqc_quality.png
new file mode 100644
index 0000000..25104c1
Binary files /dev/null and b/materials/01-intro/images/fastqc_quality.png differ
diff --git a/materials/01-intro/images/gisaid_metadata.png b/materials/01-intro/images/gisaid_metadata.png
new file mode 100644
index 0000000..49b4cfb
Binary files /dev/null and b/materials/01-intro/images/gisaid_metadata.png differ
diff --git a/materials/01-intro/images/igv_overview.svg b/materials/01-intro/images/igv_overview.svg
new file mode 100644
index 0000000..8af4d14
--- /dev/null
+++ b/materials/01-intro/images/igv_overview.svg
@@ -0,0 +1,2258 @@
+
+
+
+
diff --git a/materials/01-intro/images/lineages_example.svg b/materials/01-intro/images/lineages_example.svg
new file mode 100644
index 0000000..c503d4c
--- /dev/null
+++ b/materials/01-intro/images/lineages_example.svg
@@ -0,0 +1,4282 @@
+
+
+
+
diff --git a/materials/01-intro/images/metagenomic_vs_amplicon.svg b/materials/01-intro/images/metagenomic_vs_amplicon.svg
new file mode 100644
index 0000000..8f2e9f7
--- /dev/null
+++ b/materials/01-intro/images/metagenomic_vs_amplicon.svg
@@ -0,0 +1,819 @@
+
+
diff --git a/materials/01-intro/images/multiqc.svg b/materials/01-intro/images/multiqc.svg
new file mode 100644
index 0000000..50ab9d5
--- /dev/null
+++ b/materials/01-intro/images/multiqc.svg
@@ -0,0 +1,3435 @@
+
+
+
+
diff --git a/materials/01-intro/images/ngs_mapping.svg b/materials/01-intro/images/ngs_mapping.svg
new file mode 100644
index 0000000..9c10e65
--- /dev/null
+++ b/materials/01-intro/images/ngs_mapping.svg
@@ -0,0 +1,509 @@
+
+
+
+
diff --git a/materials/01-intro/images/virus-RT-qPCR.png b/materials/01-intro/images/virus-RT-qPCR.png
new file mode 100644
index 0000000..166939b
Binary files /dev/null and b/materials/01-intro/images/virus-RT-qPCR.png differ
diff --git a/materials/02-isolates/01-consensus.html b/materials/02-isolates/01-consensus.html
new file mode 100644
index 0000000..88f61cd
--- /dev/null
+++ b/materials/02-isolates/01-consensus.html
@@ -0,0 +1,1127 @@
+
+
+
+
+
+
+
+
+
+SARS Genomic Surveillance
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
4Consensus assembly
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Learning Objectives
+
+
+
+
+
Recognise what the main steps are in processing raw sequencing data to generate consensus genome sequences, including sequence alignment, primer trimming and consensus generation.
+
Recognise the differences between Illumina and Nanopore pipelines.
+
Apply the nf-core/viralreconNextflow pipeline to generate a consensus sequence from Illumina and Nanopore data.
+
+
+
+
+
+
4.1 SARS-CoV-2 Consensus Assembly
+
As we discussed earlier in the course, the starting material for sequencing SARS-CoV-2 samples from infected patients is PCR-amplified DNA generated with a panel of primers that covers the whole SARS-CoV-2 genome (for example the primers developed by the ARTIC network). This material can then be sequenced using either Illumina or Nanopore platforms.
+
Although different sotware tools are used depending on which kind of sequencing platform was used, the main goal is the same: to align the sequencing reads to the reference genome, and identify any DNA changes (SNPs or Indels) relative to the reference genome (Wuhan-Hu-1). This is called consensus assembly, since we are assembling the genome of our sample from the PCR-amplified fragments and generating a consensus sequence based on changes present in several reads covering a particular position of the genome.
+
The general data processing steps are:
+
+
Filter high-quality sequencing reads.
+
Map the reads to the Wuhan-Hu-1 reference genome.
+
Trim the primers from the aligned reads based on the primer location file (BED file).
+
Perform variant calling (SNPs and indels) to identify changes relative to the reference sequence.
+
Generate a consensus sequence for the sample based on those variants.
+
+
+
+
+Overview of the consensus assembly procedure from amplicon sequencing reads. In this schematic, each read spans the whole length of a PCR amplicon, which is what is expected from Nanopore reads. With Illumina data, there would be two pairs of reads starting at each end of the PCR amplicon.
+
+
+
+
+
+
+
+
+Primer trimming
+
+
+
+
Primer trimming is a key step of the data processing, otherwise SNPs might be missed at the primer sites, on the final consensus sequence. This is because the primer sequence is retained during PCR instead of the original sequence of the sample. Because the PCR amplicons overlap with each other, we can trim the primers from each read and do variant calling after trimming. An example of this is shown in the Figure above.
+
+
+
+
+
+
4.2 Bioinformatic Workflows/Pipelines
+
As can already be seen from the brief description above, bioinformatic analyses always involve multiple steps where data is gathered, cleaned and integrated to give a final set of processed files of interest to the user. These sequences of steps are called a workflow or pipeline. As analyses become more complex, pipelines may include the use of many different software tools, each requiring a specific set of inputs and options to be defined. Furthermore, as we want to chain multiple tools together, the inputs of one tool may be the output of another, which can become challenging to manage.
+
Although it is possible to code such workflows using shell scripts, these often don’t scale well across different users and compute setups. To overcome these limitations, dedicated workflow/pipeline management software packages have been developed to help standardise pipelines and make it easier for the user to process their data.
+
+
Two of the most popular workflow software packages are Snakemake and Nextflow. We will not cover how to develop workflows with these packages, but rather how to use an existing workflow to generate consensus sequences from SARS-CoV-2 data.
+
+
Why Use a Standardised Workflow?
+
These are some of the key advantages of using a standardised workflow for our analysis:
+
+
Fewer errors - because the workflow automates the process of managing input/output files, there are less chances for errors or bugs in the code to occur.
+
Consistency and reproducibility - analysis ran by different people should result in the same output, regardless of their computational setup.
+
Software installation - all software dependencies are automatically installed for the user using solutions such as Conda, Docker and Singularity (more about these in a later section of the course).
+
Scalability - workflows can run on a local desktop or scale up to run on high performance compute clusters.
+
Checkpoint and resume - if a workflow fails in one of the tasks, it can be resumed at a later time.
+
+
+
+
+
4.3 SARS-CoV-2 Pipeline
+
To generate consensus SARS-CoV-2 genomes from these data, we will use a pipeline that was developed by the Nextflow core team called nf-core/viralrecon (which was itself inspired by a previous pipeline from the Connor Lab). Its objective is to harmonise the assembly of SARS-CoV-2 genomes from both Illumina and Nanopore amplicon sequencing data. It can also work with metagenomic data, which we will not cover in this workshop. This pipeline therefore includes different sub-pipelines, which are launched depending on the type of sequence data we have. Watch the video below to learn more about the development of this pipeline.
+
+
+
+
Generally speaking, Nextflow pipelines are run with the command nextflow run PIPELINE_NAME, where “PIPELINE_NAME” is the name of the pipeline. Pipelines are usually shared in a public repository such as GitHub, and nextflow will automatically download the pipeline if it hasn’t been downloaded already to your computer.
+
Let’s test our pipeline by looking at its help documentation:
+
nextflow run nf-core/viralrecon -r 2.6.0 --help
+
The command should print a long list of options available with this pipeline. For pipelines developed by the Nextflow core team you can also consult the documentation available online, which is easier to read: nf-co.re/viralrecon. This page includes many details about the pipeline: which tools are used in different steps of the data processing, how to use the pipeline for different types of data, a detailed documentation of all the options of the pipeline and explanation of the output files generated by it.
+
Below, we give an overview of the pipelines used for Illumina and Nanopore amplicon data.
+
+
+
+
+
+
+Reference Genome and Primer Locations
+
+
+
+
The Wuhan-Hu-1 reference genome sequence and the amplicon primer locations (in BED file format) can all be found on the ARTIC Primer Schemes repository. The pipeline we are using takes care of downloading these files for us automatically, however it can be useful to know where to find them, in case you want to use other tools that require these files.
The Illumina sub-workflow is based on several standard bioinformatic tools and, importantly, on the iVar software, which was developed for analysing amplicon-based sequencing data.
+
+
+
+Schematic of the key steps in the Illumina sub-workflow.
+
+
+
To run the pipeline on Illumina data, we use the following general command:
One of the key options is --platform illumina, which makes sure that the correct sub-workflow will be used.
+
+
+Click to see more details about this sub-workflow
+
+
In summary, the steps performed by the Illumina sub-workflow are:
+
+
Adapter trimming - this consists of trimming (or “cutting”) the sequences to remove low-quality bases and any Illumina adapter sequences that are present in the sequences.
+
Removing human (host) reads - when doing the sequencing it is possible that many reads are still from human material and this step removes them from the rest of the analysis.
+
Read mapping - aligning (or mapping) the reads to the Wuhan-Hu-1 reference genome.
+
+
The software used for mapping is bowtie2.
+
The software samtools is used to convert the mapped file to BAM (instead of SAM) and sort the reads by coordinate (which is necessary for downstream steps).
+
+
Trim Primers - primers are removed from the aligned reads using ivar trim (using the primer BED file).
+
Call variants - identify SNPs and indels using ivar variants.
+
Annotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.
+
Make consensus - apply the SNPs and indels from the previous step to the reference FASTA file.
+
+
There are two tools that can be used in this step: bcftools consensus (default) or ivar consensus (can be set with the option --consensus_caller ivar).
+
+
Lineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.
+
Quality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.
This sub-workflow is similar to the other nanopore sub-workflow, the main difference is the software used for generating a consensus sequence (medaka instead of nanopolish).
+
+
+
+Schematic of the key steps in the Medaka sub-workflow.
+
+
+
To run our pipeline on basecalled data (FASTQ files), we use the following command:
--platform nanopore makes sure that the correct sub-workflow will be used.
+
--artic_minion_caller medaka indicates we want to use the medaka program to do the variant/consensus calling (directly from the basecalled FASTQ files, rather than from the raw signal in the FAST5 files).
+
--artic_minion_medaka_model specifies the model used by the guppy_basecaller software to do the basecalling. The model name follows the structure {pore}_{device}_{caller variant}_{caller version}. See more details about this in the medaka models documentation. Note: for recent versions of Guppy (>6) there is no exact matching model from medaka. The recommendation is to use the model for the latest version available; a list of supported models can be found on the medaka GitHub repository.
+
--fastq_dir specifies the directory containing the FASTQ files. This directory should contain sub-directories for each barcoded sample following the naming convention barcodeXXXX (where X is a number between 0 and 9). By default, the guppy_basecaller software from Nanopore generates a folder called “fastq_pass” which follows this convention.
+
+
+
+Click to see more details about this sub-workflow
+
+
In summary, the steps performed by the Medaka sub-workflow are:
+
+
Aggregate reads from each sequencing barcode (when multiple files are availble for each barcode)
+
Run the artic minion tool, which internally does several steps:
+
+
Map reads to the reference genome using minimap2 (can be changed to use bwa mem with the option --artic_minion_aligner bwa).
+
Trim primers from the aligned reads based on the known primer positions in the BED file (using a custom python script called align_trim.py).
+
Call consensus sequences and SNP/indel variants using medaka consensus and medaka variant:
+
+
Positions with less than 20x depth are treated as missing data and converted to the ambiguous base ‘N’. It is not advised to go below this threshold as the models used to call variants do not perform as well.
+
+
+
Annotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.
+
Lineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.
+
Quality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.
This sub-workflow is similar to the other nanopore sub-workflow, the main difference is the software used for generating a consensus sequence (nanopolish instead of medaka).
+
+
+
+Schematic of the key steps in the Nanopolish sub-workflow. (Under development)
+
+
+
+
+Click to see more details about this sub-workflow
+
+
In summary, the steps performed by the Nanopolish sub-workflow are:
+
+
Filter reads to ensure they pass minimum read length thresholds:
+
+
minimum length 400bp (can be changed with --min_length option)
+
maximum length 700bp (can be changed with --max_length option)
+
+
Run the artic minion tool, which internally does:
+
+
Read alignment to reference genome using minimap2 (can be changed to use bwa mem with the --bwa option).
+
Trim primers from the aligned reads (based on the known primer positions in the BED file).
+
Call consensus sequences and variants using nanopolish variants if using signal-level FAST5 files.
+
+
Positions with less than 20x depth are assigned the ambiguous base ‘N’. It is not advised to go below this threshold as the models used to call variants do not perform as well.
+
+
+
Annotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.
+
Lineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.
+
Quality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.
+
+
+
To run our pipeline on signal-level data (FAST5 files), we use the following command:
--platform nanopore makes sure that the correct sub-workflow will be used.
+
--fastq_dir specifies the directory containing the FASTQ files generated by the guppy_basecaller program (this is the standard software from Nanopore that processes the raw signal data from the sequencing device). This directory should contain sub-directories for each barcoded sample following the naming convention barcodeXXXX (where X is a number between 0 and 9). By default, guppy_basecaller generates a folder called “fastq_pass” which follows this convention.
+
--fast5_dir specifies the directory containing the FAST5 files generated by guppy_basecaller. This directory follows the same naming convention as above and is usually in a folder called “fast5_pass”.
+
--sequencing_summary is a path to the “sequencing_summary.txt” text file generated by guppy_basecaller.
+
+
+
+
+
Apart from the specific options used by each sub-workflow, there are some general options that are used:
+
+
--input specifies a CSV file with details about our samples. The format of this file depends on the specific sub-workflow you are using. See the details in the samplesheet documentation page.
+
--outdir specifies the output directory to store all our results.
+
--protocol amplicon sets the pipeline for PCR amplicon data (the other option is --protocol metagenomic, which we do not cover in this course).
--primer_set artic at the moment only “artic” primers are available by default. It is possible to use custom primers with the Illumina workflow (see details here).
+
--primer_set_version the version of the ARTIC primer scheme used. The viralrecon primer config file indicates the available primer shemes are: 1, 2, 3, 4, 4.1, 5.3.2 and also 1200 (the 1200bp amplicon protocol, also known as “midnight”).
+
--skip_assembly this is used to skip de-novo assembly of the genome. This step is unnecessary in amplicon protocols, which instead rely on mapping reads to the reference genome (reference-based assembly). De-novo assembly is necessary for metagenomic protocols.
+
--skip_pangolin and --skip_nextclade is used to skip the lineage assignment. The reason is that viralrecon does not use the latest version of the SARS lineage databases, so we skip this step for now, and run it separately in a later step of the analysis.
+
+
There are two more generic options we used:
+
+
-r 2.6.0 indicates the version of the pipeline we want to use. It’s always good to check what the latest version is on the viralrecon website. -profile singularity indicates how we want to manage the software required by the pipeline. In our case, we are using a software called Singularity, which creates a “virtual operating system” (called a container) where all the necessary software is run from. This ensures that all of the software is automatically installed and runs on any Linux computer.
+
+
+
+
+
+
+
+Conda, Singularity, Docker?
+
+
+
+
Generally speaking, workflow management software such as Nextflow or Snakemake support three solutions for managing software dependencies:
+
+
Docker is a software that allows to package a small virtual operating system (or a “container”) containing all the software and data necessary for running an analysis.
+
Singularity also creates software containers, similarly to Docker. However, it can more easily interface with the user’s filesystem, without the need to have special permissions.
+
Conda is a package manager, also very popular in bioinformatics. Instead of creating virtual OS containers, Conda instead creates software environments (think of them as directories) where all the software is locally installed, including all its dependencies. The use of individual environments ensures that software packages with incompatible dependencies can still be used within the same pipeline.
+
+
Of the three, Singularity is the recommended choice, although Conda is also a good alternative.
+
+
+
+
4.3.1 Running the Workflow
+
Let’s see an example in action by using some example data. If you go to the directory uk_illumina/ in the course materials, you will find several FASTQ files in the data directory. There is also a shell script (scripts/run_illumina_workflow.sh) that contains the commands we will use to run the workflow on these data.
+
Opening the script, we can see the following commands:
It first creates a results directory (to store our output files) and then runs the nextflow command using the Illumina sub-workflow. We could run these commands one at a time by copy/pasting them to the terminal. Or alternatively, we can run the entire script using bash scripts/run_illumina_workflow.sh
+
When you start running the workflow, you will get a list of the workflow steps and their progress. This may take quite a while to run, depending on the computer resources you have available. Once the workflow is complete, you should see a message similar to the following:
You should also get several output files in the results folder specified with our nextflow command. We will detail what these files are in the next section.
+
+
+
+
+
+
+Running the pipeline on our training computers
+
+
+
+
Our training computers don’t have the high specifications needed for routine bioinformatic analysis, so the Illumina pipeline takes up to 1h to complete.
+
We provide already pre-processed results for 48 samples in the folder uk_illumina/preprocessed, which you can use to follow the next section.
+
+
+
+
+
+
4.4 Exercises
+
+
+
+
+
+
+Running nf-core/viralrecon: ONT data
+
+
+
+
+
+
+
+
Go to the course materials directory india_nanopore. This contains Nanopore sequencing data for several samples collected in India. Nanopore data is organised in directories named as barcodeXX (where XX is a number) - this is the standard output from the Guppy software used to do basecalling and generate FASTQ files.
+
The Nanopore Medaka workflow expects to be given as an input a CSV file with two columns: sample name and barcode number. We already provide this file in samplesheet.csv.
+
Your task now is to process these samples using the nf-core/viralrecon pipeline:
+
+
Using nano, open the script found in scripts/run_medaka_workflow.sh.
+
Fix the code in the script where you see the word “FIXME”:
+
+
Output the results to a directory called results/viralrecon/.
+
The input sample sheet is in the file samplesheet.csv (check the pipeline documentation to review what the format of this samplesheet should be for the Nanopore pipeline).
+
The FASTQ files are in a folder data/fastq_pass.
+
+
Run the script using bash. This may take ~15 minutes to complete.
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
We can open the script with Nano using the command:
Set --input as samplesheet.csv, which is the CSV file with two columns: sample name and Nanopore barcode number. The format of this file is detailed in the nf-core/viralrecon documentation.
+
Set --outdir as results/viralrecon/, which is the directory where we want to output our results.
+
Set --fastq_dir as data/fastq_pass, which is the directory containing the FASTQ file folders named as barcodeXX (where XX is a number). This is the standard output from the Guppy software used to do basecalling of Nanopore data to produce FASTQ files.
+
+
When we finish editing the file we can hit Ctrl + X to exit Nano. Before it closes it will ask us if we want to save the file and we can type “Y” for yes.
+
To run the script we can do bash scripts/run_medaka_workflow.sh. After the workflow completes, we get a message similar to this one:
Note that the exact time that the workflow takes to run may differ from what we show here. The time depends on how many samples you are processing and how big the computer you are using is.
+
+
+
+
+
+
+
+
+
+
+
+
+
4.5 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
The main steps to generate SARS-CoV-2 consensus sequences are: filter high-quality reads, map reads to reference genome, trim PCR primers and variant/mutation calling, which is finally used to produce a consensus sequence.
+
Other steps that can be done include annotating the variants/mutations (what their effects in each gene might be) and assigning sequencences to known lineages/clades.
+
Nextflow is a software used for building workflows/pipelines involving multiple tools and data processing steps. Using established pipelines helps with automation, reproducibility, consistency of results and reduces the chances of data processing errors.
+
The nf-core/viralrecon pipeline implements the steps to generate SARS-CoV-2 consensus sequences from Illumina or Nanopore data.
+
The command nextflow run nf-core/viralrecon is used to run the pipeline, using specific options depending on the data we have.
Interpret and critically evaluate data quality reports from the assembled sequences and identify sequences suitable for downstream analyses.
+
+
+
+
+
5.1 Output Files
+
After running our pipeline, we get several output directories and files. The directories we get depend on which version of the workflow was used (Illumina or Nanopore). The description of the output is detailed in the pipeline documentation. Although there are many output files, most of these contain results that are aggregated in an interactive MultiQC report, which makes their analysis easier. We highlight some of the main files of interest below.
The file multiqc/multiqc_report.html contains a MultiQC quality and analysis report for the consensus assemblies generated by the pipeline.
+
The folder variants/bowtie2/ contains individual BAM files, which can be visualised with IGV if we want to look at the reads mapped to the reference genome.
+
The folder variants/ivar/consensus/bcftools contains individual FASTA files (named *.consensus.fa) for each sample’s consensus genome sequence.
+
The file variants/ivar/variants_long_table.csv contains a table with the aggregated results of all the variants detected in each sample.
+
+
+
+
+
The file multiqc/medaka/multiqc_report.html contains a MultiQC quality and analysis report for the consensus assemblies generated by the pipeline.
+
The folder medaka/ contains:
+
+
Individual BAM files (named *.primertrimmed.rg.sorted.bam), which can be visualised with IGV if we want to look at the reads mapped to the reference genome.
+
Individual FASTA files (named *.consensus.fasta) for each sample’s consensus genome sequence.
+
A file called variants_long_table.csv with a table of all the variants detected in each sample.
+
+
+
+
+
+
+
+
5.2 Quality Control
+
The viralrecon pipeline produces many quality control metrics, which are conveniently compiled in an interactive report with MultiQC, as mentioned above. We will not detail here every section of the report (check the pipeline documentation for a full description), but only highlight some of the sections that can be used for a first assessment of the quality of our samples.
+
+
5.2.1 Variant Calling Metrics
+
The first section or the report - Variant Calling Metrics - contains a summary table with several statistics for each sample, including the total number of reads, the number/percentage of reads mapped to the reference genome, the depth of coverage, the number of SNPs (single-nucleotide polymorphisms) and INDELs (insertions/deletions), the number of missense variants (mutations that result in an aminoacid change) and the fraction of ambiguous bases ‘N’ per 100kb.
+
Looking at these basic metrics gives us a good first idea of whether some of the samples have a high fraction of missing bases (marked as N in the FASTA file), leading to a poorer assembly. We can quickly check this by sorting the table by the column named “# Ns per 100kb consensus” (you can convert the values in this column to a percentage by dividing the numbers by 100). There is also the ability to produce simple scatterplots from the data in this table, which can be useful to look at the relationship between the different metrics (see an example in the figure below).
+
This table also contains information about the lineage/clade assigned to each sample by the programs Pangolin and Nextclade. This gives us an idea of which samples may be more similar to each other, and where they fit in the global context of other sequences available publicly. We will talk more about this topic in the lineage assignment section.
+
+
+
+
+Snapshot of the “Variant Metrics” section of the viralreconMultiQC report. Simple scatterplots can be made from the data on this table using the Plot button. For example, at the bottom we show a scatterplot showing the relationship between the median depth of coverage and the number of ambiguous bases ‘N’ per 100kb. For the data in this example, we can see that when the average depth of coverage drops below around 200 reads we start getting higher number of missing bases in the assembly.
+
+
+
+
+
+
+
+
+Terminology Alert!
+
+
+
+
The word coverage is sometimes used in an ambiguous way by bioinformaticians. It can mean two things:
+
+
The number of reads aligning to a position of the genome. This is sometimes called sequencing depth or depth of coverage (we will use these terms in the materials to avoid confusion). For example, we may say that the average depth of coverage of the genome is 20x, meaning that on average there are 20 reads aligned to a position of the genome.
+
The fraction of a genome that has a sufficient number of reads for analysis. For example, if 90% of the genome has a sufficient number of reads to be analysed (for example, at a threshold of 10x), we would say it has 90% coverage (we “covered” 90% of the genome with enough data). In the context of genome assembly, we sometimes use the word “coverage” to refer to the percentage of the consensus genome without ambiguous ‘N’ bases.
+
+
In the viralreconMultiQC report the word “coverage” is used to mean “depth of coverage”.
+
+
+
+
+
5.2.2 Amplicon Depth of Coverage
+
The next section of the report - Amplicon Coverage Heatmap - contains a graphical representation of the average depth of coverage for each PCR amplicon in the ARTIC protocol (i.e. the average number of reads mapped to each amplicon interval). This plot is extremely useful to identify common PCR dropouts occurring across many samples. This may be an indication that our PCR primer scheme is not working for some of the forms of the virus circulating in our population (for example due to mutations that accumulate in the primer site).
+
+
+
+Example heatmap showing the depth of coverage for a set of samples. The failure of an amplicon to be sequenced in several samples can be seen as a “column” of dark cells in the heatmap.
+
+
+
We can investigate primer dropout in more detail, for example by looking at the BAM files with mapped reads using IGV.
+
From our heatmap (shown in the Figure above) we can see one of the PCR fragments - ncov-2019_83 - seems to have poor amplification across many of our samples. Let’s investigate this in more detail by looking at the alignment file:
+
+
Open IGV and go to File → Load from file….
+
In the file browser that opens go to the folder results/viralrecon/variants/bowtie2/ and select the file GB36.ivar_trim.sorted.bam to open it.
+
Go back to File → Load from file… and this time load the BED files containing the primer locations. These can be found in resources/primers/artic_primers_pool1.bed and resources/primers/artic_primers_pool2.bed.
+
+
Once you have IGV open, you can navigate to the region where this amplicon is located by searching for the name of one of the primers - “ncov-2019_83_LEFT” - in the search box at the top. By zooming in to the region where this primer is located, we can see there is a mutation right at the start of this primer, which suggests that this may be the reason why this PCR fragment fails to amplify in this sample.
+
+
+
+Screenshot from the IGV program showing an example of a region with PCR amplicon dropout. This is shown by a lack of reads mapped to this region of the genome. Because the PCR fragments from the ARTIC protocol overlap between the two pools, we can see if there are mutations occurring at the primer sites. In this example, a mutation in the left primer of amplicon 83 suggests this may be the reason for the dropout in this sample.
+
+
+
+
+
+
5.3 Mutation/Variant Analysis
+
One of the output files produced by the pipeline is a table SNP and indel variants detected in our samples saved as a CSV file in variants/ivar/variants_long_table.csv. This table can be useful to investigate if particular mutations are particularly frequent in our samples and what their likely effects are.
+
+
+
+Variants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases.
+
+
+
The columns in this table are:
+
+
SAMPLE is the name of our sequenced sample.
+
CHROM is the name of the “chromosome”, which in our case is just the name of the reference genome (“MN908947.3”).
+
POS is the position of the genome where the mutation occurs.
+
REF is the reference nucleotide.
+
ALT is the alternative nucleotide, that is the nucleotide that was observed in our sample.
+
FILTER indicates whether the SNP passed quality filters from the variant calling software (“PASS”) or whether some other issue was observed (for example “dp” means that the depth of sequencing was unusually high or low).
+
DP is the total depth of sequencing at this position, meaning how many reads in total were aligned there.
+
REF_DP is the depth of sequencing of the reference allele, meaning how many reads contained the reference nucleotide.
+
ALT_DP is the depth of sequencing of the alternative allele, meaning how many reads contained the alternative nucleotide.
+
AF is the allele frequence of the alternative allele, meaning the proportion of reads that contained the alternative nucleotide (this column is equivalent to ALT_DP/(ALT_DP + REF_DP)).
+
GENE is the name of the gene in the SARS-CoV-2 annotation.
+
EFFECT this is the predicted effect of the mutation in the gene. This output comes from the snpeff software and uses The Sequence Ontology nomenclature (follow the link to search for each term).
+
HGVS_C, HGVS_P and HGVS_P_1LETTER is the DNA or amino acid change using HGVS nomenclature. For example, “c.1112C>T” means a C changed to a T at position 1112 of the genome; and “p.Pro371Leu” would mean that a Proline changed to a Leucine at position 371 of the respective protein.
+
CALLER is the software used for variant calling.
+
LINEAGE is the Pangolin lineage that the sample was assigned to. Note that this column should usually be ignored, since viralrecon doesn’t use the latest Pangolin data version by default.
+
+
This table can be opened in a spreadsheet program such as Excel to look at particular features. In terms of quality-control, we can filter the table:
+
+
for mutations with intermediate allele frequency (say < 70%) – if a sample has too many such mutations, that could indicate cross-contamination between samples.
+
for frameshift mutations – these mutations should be rare because they are highly disruptive to the functioning of the virus, and their occurrence is more often due to errors. The presence of these mutations are usually not critical for downstream analysis (such as lineage assignment and phylogenetics), but we should make a note that these mutations may be false-positives in our data.
+
+
This variants/mutations table can also be used to explore reasons for amplicon dropout. For example, we can identify how many samples contain the mutation we previously saw in the “ncov-2019_83_LEFT” primer (we saw this was in position 25,003 of the genome). We can do this by sorting our table by the column “POS” (position) and then scrolling down to find the samples with this mutation. We will find that only 3 of the samples contain this mutation, suggesting some other additional causes are leading to the dropout in this PCR fragment.
+
Finally, for the Illumina pipeline (--platform illumina), it is important to note that not all of the mutations on this table are included in the final consensus. Only mutations with allele frequency >75% (AF >= 0.75) and minimum depth of 10 (DP >= 10) are retained in the final consensus. Therefore, to see which mutations are actually present in our consensus sequences, we need to filter this table for these criteria. Again, we note that this only applies to the Illumina pipeline. For the Nanopore pipeline (--platform nanopore) all the mutations included in this table are retained in the consensus sequence.
+
+
+
+
+
+
+ORF1b Annotation Issues
+
+
+
+
The annotation of the ORF1b gene had some changes between the first original assembly and newer versions of this annotation. In particular, there is a frameshift that is not considered in the annotation used by most software, and this causes a discrepancy in the results between viralrecon and other tools such as Nextclade (which we will cover in the next section).
+
This issue is under investigation by the developers of viralrecon and may be corrected in future versions of the pipeline.
+
For now, the advice is to ignore the variant effects of ORF1b as these correspond to an older version of the annotated gene.
+
+
+
+
+
+
+
+
+Keep Original Files Intact
+
+
+
+
When you analyse the results of a CSV file in a spreadsheet software (such as Excel), it may be a good idea to create a copy of your file to avoid accidentally modifying the original table. For example, if you accidentally sort one column of the table but forget to sort other columns, of if you accidentally filter the results and delete some of the rows from the table, etc.
+
You can save a copy of the file by going to File → Save As…. You may want to save the copy as an Excel file format, to include graphs and colours. (Remember that the CSV format is a plain text file - it does not support graphs or coloured cells.)
+
+
+
+
+
5.4 Cleaning FASTA Files (Optional)
+
To proceed with our analysis, we need a FASTA file containing all of our consensus sequences. However, our viralrecon pipeline outputs separate FASTA files for each sample. We can see this by running (from within the uk_illumina/ directory):
+
ls results/viralrecon/variants/ivar/consensus/bcftools/
+
Also, the workflow modifies our original sample names in the FASTA file, by adding extra information to the sequence name. For example:
What we want to do is clean these sample names, so that we end up with:
+
>ERR5728910
+
We also want to make sure to combine all the samples into a single FASTA file.
+
We can the command cat to combine (concatenate) the individual files and the sed command to substitute text and clean our sample names. Let’s do this step by step.
+
First, we can use the *wildcard to combine all the FASTA files with the cat command:
Running this command will print all of the sequences on the screen! To see what happened a little better, we could pipe this command to less to browse up-and-down through the file:
We could also check that we now have all our samples combined, we could pass the results to grep and search for the word >, which in the FASTA format indicates the sequence name:
Notice that in this last command we make sure to redirect the result to a new file using >.
+
+
+
5.5 Missing data intervals
+
When we align reads to the reference genome, there may be regions that we were not able to sequence (e.g. due to amplicon dropout, discussed above) and therefore cannot tell what the base sequence in those positions is. In those cases, the viralrecon pipeline includes the missing character ‘N’ in the sequence.
+
As a further quality check, it is useful to check how many contiguous intervals of missing bases we have in each of our assemblies, as well as how long those intervals are. For example, for this small sequence:
+
T A N N N G C T N N A T
+
We have two missing intervals, from positions 3-5 and from positions 9-10.
+
To obtain a list of missing intervals in a sequence, we can use the software seqkit, which is a toolkit of commands to do many operations on FASTA/FASTQ files (check out its documentation). In particular, we can use the following command:
The output is a tabular file specifying the consensus sequence name (first column) and the location of intervals in the genome where the pattern of one or more “N” were found.
+
To see what all the options we used with this command are, see the tool’s documentation.
+
+
+
5.6 Exercises
+
+
+
+
+
+
+Quality control for India (ONT) samples
+
+
+
+
+
+
+
+
Now it’s your turn to check the quality of the assembled genomes from our India samples, sequenced using Nanopore. For this exercise, we will use the results produced by viralrecon on 48 pre-processed samples:
+
+
Use the file explorer to open the folder preprocessed, and then:
+
+
Open the file in pipeline_info/execution_report_DATE.html (where “DATE” is the date when the files were processed).
+
How long did the workflow take to run?
+
Which step of the pipeline took the longest to run?
+
+
Open the MultiQC report found in preprocessed/multiqc/medaka/multiqc_report.html and answer the following questions:
+
+
Produce a plot between median depth of coverage (x-axis) and #Ns (y-axis). What do you think is the average coverage needed for <10% of missing data.
+
Were there any PCR amplicons with a dropout (i.e. very low depth of coverage) across multiple samples?
+
Do any of these dropout amplicons coincide with gene “S” (Spike protein gene)? Note: you can see the ARTIC PCR primer location from the online repository and the gene locations in the SARS-CoV-2 NCBI genome browser.
+
+
Based on any amplicons that you identified in the previous question, find if there are any mutations coinciding with their primer locations. You can use the mutation variant table in preprocessed/medaka/variants_long_table.csv.
+
With the mutation variants table open in a spreadsheet program (LibreOffice on our training machines):
+
+
Use the filter button to look for mutations in gene “S” (encodes for the Spike protein).
+
Identify the samples and positions of mutations of the type “disruptive_inframe_deletion”.
+
Using IGV, open the BAM file for one of those samples. Note: BAM files are located in preprocessed/medaka/SAMPLE.primertrimmed.rg.sorted.bam (were ‘SAMPLE’ is the sample name).
+
Go to the location of those mutations. From the read alignment, how confident are you about it? What could you do to confirm it?
+
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
Question 1
+
We open the file found in preprocessed/pipeline_info/execution_report_2022-05-04_12-41-12.html, which contains information about the pipeline that was run on a set of 48 samples. We can see at the top of the report that it took ~15 minutes to run. This report also gives us information about how long each individual step of the pipeline took the longest to run (and other information such as which used more CPU or RAM memory). For example, in the section “Job Duration” we can see a graph that looks like this:
+
+
This indicates that the step running the artic minion tool takes the longest. This is not surprising as this is the step where most of the work is happening (mapping, primer trimming and making a consensus sequence). You can revise the steps of the workflow in the respective section above.
+
+
Question 2
+
When opening the MultiQC report, we can see the first section contains a table with several statistics about the quality of each sample. We can produce a plot from this table, by pressing the “Plot” button above the table. The plot we were asked for is the following:
+
+
+
+This plot can be produced by clicking the Plot button on top of the table and then selecting the variables “Coverage median” and “# Ns per 100kb consensus”.
+
+
+
The y-axis of the plot tells us the number of ‘N’ per 100kb of sequence. If we divide those numbers by 100 we get a percentage and from these samples it seems like a median depth of coverage of around 200 reads generates samples with less than 10% of missing bases.
+
From the section of the report “Amplicon coverage heatmap” we can see the average depth of coverage for each PCR amplicon from the ARTIC protocol. There are 3 amplicons in particular that have very low depth of coverage: nCoV-2019_51, nCoV-2019_62 and nCoV-2019_95.
+
We can check the ARTIC V3 primer BED file online to look at the location of these primers, or we could look for it using command-line tools. Here is an example:
For this example, we can see the amplicon is between positions 15171 - 15560 of the genome. Looking at the SARS-CoV-2 NCBI genome browser, we can see this coincides with ORF1ab. If we do the same analysis for the other primers, we will see that none of them coincides with gene S.
+
+
Question 3
+
To investigate whether this amplicon dropout is due to mutations in the primer regions we can open the mutations table preprocessed/medaka/variants_long_table.csv. If we sort the table by the column “POS”, we can scroll to the location of each primer pair.
+
We can see that in only one case there is a mutation coinciding one of the primers: in sample “IN33” position 28687 there is a C > T mutation, which coincides with primer nCoV-2019_95_LEFT. However, this only occurs for one of the samples, suggesting the reason for the PCR dropout might be related to other factors.
+
+
Question 4
+
After filtering our table for gene S (Spike gene), we can see only a couple of mutations of type “disruptive_inframe_deletion” in some of the samples at positions 21764 and 21990. To look at the reads with this mutation, we open the BAM file for one of these samples in IGV (for example sample IN22):
+
+
Go to File → Load from file….
+
In the file browser that opens go to the folder preprocessed/medaka and select the file IN22.primertrimmed.rg.sorted.bam to open it.
+
In the search box we can type “NC_045512.2:21990” which will zoom-in on the region around the deletion.
+
+
From looking at the reads aligned to this position of the genome, we can see several of them containing a 3bp deletion. However, we can also see some reads that do not contain the deletion, and others that seem to be mis-aligned. If we were interested in confirming this mutation, we could do an independent PCR followed by Sanger sequencing, for example.
+
Although this was not part of the question, it is also worth noting that the primer “nCoV-2019_73_LEFT” starts very near this deletion. In the MultiQC report, we can see that this PCR amplicon had very poor amplification in this sample. One possibility is that the deletion interfered with the primer efficiency in this sample.
+
Generally, we don’t need to confirm every single mutation obtained from our analysis. But if we see a mutation occurring many times, then it may be worth further investigation, especially if it is disruptive and in an important gene such as the Spike gene.
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Clean FASTA file: ONT data
+
+
+
+
+
+
+
+
In this exercise we will create a clean FASTA file for the samples collected in India.
+
In this case, the output FASTA files are in the folder results/viralrecon/medaka and have the file extension .fasta. If we look at one of the files:
We can see that the name has a lot of extra information attached to it. We want to clean the name of the sequences so that the result is:
+
>IN42
+
The following sed command can be used to substitute the text “/ARTIC/medaka MN908947.3” with nothing:
+
sed's|/ARTIC/medaka MN908947.3||'
+
+
Pipe the tools cat and sed to construct a command that generates a new file called results/clean_sequences.fa containing all the sequences with “clean” sequence names.
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
The complete code to achieve the desired outcome is:
The output of the pipeline includes, among others:
+
+
A detailed MultiQC report, including information about genome coverage, the depth of sequencing for each amplicon, as well as other quality metrics that can help to troubleshoot problematic samples.
+
A table with SNP and indel mutation variants identified in each sample.
+
Information about SARS-CoV-2 lineages/clades/variants, which is detailed in the next section.
Understand variant annotation conventions used by Gisaid, Pango, Nextstrain and WHO and how they relate to each other.
+
Assign sequences to Pango lineages using pangolin.
+
Interactively explore mutations in the assembled genomes and their phylogenetic context using Nexstrain’s tools.
+
+
+
+
+
+
6.1 SARS-CoV-2 Variants
+
As viruses (or any other organism) evolve, random DNA changes occur in the population, for example due to replication errors. Many of these changes are likely to be neutral, meaning that they do not change the characteristics of the virus in any significant way. Neutral changes tend to drift in the population, increasing or decreasing in frequency in a random way, but most likely end up disappearing due to their low starting frequency in the population.
+
On the other hand, advantageous mutations can occur, which lead to changes in the characteristics of the virus that are beneficial for its spread in the population (e.g. high transmissibility, resistance to immune response or vaccines, etc.). Such beneficial mutations will therefore experience positive selection, potentially leading to their increase in frequency in the population as they spread to more and more hosts.
+
Viruses carrying those advantageous mutations may, over time, aquire other advantageous mutations that further increase their fitness and, therefore, their frequency in the population. One way to visualise this is by looking at a phylogenetic tree showing the relationship between sequences (based on their similarity) and how groups of sequences change over time.
+
+
+
+Example of global phylogeny from the Nextstrain public server. Colours show different Nextstrain clades. (Screenshot taken Feb 2022)
+
+
+
In the figure above, which shows SARS-CoV-2 samples from across the world, we can see groups of similar sequences rapidly “expanding” at certain points in time. Such groups of sequences, which share a collection of DNA changes, are referred to as SARS-CoV-2 variants (see box below about the ambiguous meaning of this term). In an effort to understand the spread of the virus and monitor situations of increased occurrence of such variants, several groups and institutions have developed a system to classify groups of SARS-CoV-2 sequences as variants of interest and variants of concern.
In practice, there is a big overlap between these different nomenclature systems, with WHO variants having a direct match to Pango lineages and Nextstrain clades. In fact, the different teams work together to try and harmonise the nomenclature used, and thus facilitate the interpretation of sequence analysis.
+
The two most popular systems - Nextclade and Pangolin - have slightly different levels of resolution. Nextclade’s nomenclature system was developed to highlight diversity patterns at a larger scale, allowing discussions of SARS-CoV-2 diversity at a global level and over larger time scales. On the other hand, Pangolin’s nomenclature system is more fine-grained, aiming to follow the dynamics of the pandemic as it unfolds. The two systems are complementary to each other, and our analysis of SARS-CoV-2 sequences should include both tools.
+
+
+
+
+
+
+What is a variant?
+
+
+
+
It is important to note that the term variant can be sometimes ambiguous.
+
The term “SARS-CoV-2 variant” usually refers to the WHO definition of variants of concern/interest (e.g. the Alpha, Delta and Omicron variants), which includes sequences containing a collection of several nucleotide changes that characterise that group. According to this definition, we have two variants in the example below (samples 1 & 2 are one variant and samples 3 & 4 another variant).
+
+
However, in bioinformatic sequence analysis, a sequence variant refers to an individual change in the DNA sequence (a SNP or an insertion/deletion). Using this definition, in the example above we have 5 variants: 3 SNPs and 2 indels. In the Consensus Sequence section, we saw that one of our workflow steps was “variant calling”. This was the definition of variant we were using: identifying individual SNPs and/or indels relative to the reference genome, from our sequencing data. This is also reflected in the one of the common file formats used to store SNP/indel information, the VCF file, which means “Variant Call Format”.
Because of this ambiguity, the terms “lineages” or “clades” are often used instead of “variants” when referring to groups of similar SARS-CoV-2 sequences, because they have a phylogenetic interpretation.
+
+
+
+
+
6.2 Pangolin
+
+
The first tool we will cover is called pangolin and uses the Pango nomenclature system. The main steps performed by this tool are:
+
+
Multiple sequence alignment of our samples against the Wuhan-Hu-1 reference genome, using the minimap2 software.
+
Assigning our sequences to lineages based on the current global phylogeny. Two methods/software are available:
+
+
pangoLEARN (default) uses a pre-trained machine learning model.
+
UShER uses a more classic parsimony-based method, but highly optimised for working with large numbers of sequences.
+
+
Classifying our sequences according to the WHO nomenclature of variants of interest/concern using the scorpio software.
+
+
Although Pangolin can run as part of the nf-core/viralrecon pipeline we used, we recommended to turn this option off. The reason is that the model to classify variants regularly changes over time, as more public sequences become available and the nomenclature rules updated. Therefore, it important to always run the samples through the latest Pangolin version available.
+
Pangolin can be run from the command line, using two steps:
+
+
Updating the data used for lineage/variant classification.
+
Running the actual lineage assignment step.
+
+
On our example data, these would be the commands:
+
# update pangolin data
+pangolin--update-data
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv results/clean_sequences.fa
+
+
The first command downloads the latest version of the lineages and their characteristic mutations from the Pango classification system.
+
The second command runs the FASTA file of consensus genomes through Pangolin’s classification algorithm.
+
+
--outdir is used to define the directory to save the results in.
+
--outfile is used to give the file a name of our choice.
+
+
+
The output is a CSV file, with several columns of interest, including WHO variants of concern identified using the Scorpio software. A detailed explanation of the columns of this file is given in the Pangolin documentation page.
+
More information about running Pangolin from the command line can be found in its online documentation.
+
+
6.2.1 Web Application
+
This tool can also be run separately using a web application, which only requires us to provide with a FASTA file of consensus sequences. This may desirable to re-run samples using the latest version of the Pangolin software and SARS-CoV-2 variant databases.
+
The results from the web application can be downloaded as a CSV file, which contains a table similar to the one obtained from our command above (some of the column names are different, but their meaning is the same).
+
+
+
+
6.3 Nextclade
+
+
Another system of clade assignment is provided by nextclade, which is part of the broader software ecosystem Nextstrain.
+
Nextclade performs similar steps to Pangolin, with some differences in the algorithms that are used:
+
+
Each sequence is aligned with the Wuhan-Hu-1 reference genome using a local alignment algorithm.
+
Samples are placed in the global phylogeny using a distance-based metric (placing the sequence on the tree where it has the highest similarity with).
+
Clade assignment is done based on the previous phylogeny placement step.
+
+
You can find more details about Nextclade’s methods on its documentation. Nextclade also provides several quality control metrics, which are very useful to identify problematic samples.
+
As we discussed above, the models and clades are regularly updated, so we also skipped this step when we ran the nf-core/viralrecon pipeline. Instead, we can run this tool directly from the command line, by first making sure to download the most up-to-date clade information. Here are the commands:
+
# get nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade results/clean_consensus.fa
+
+
The first command (nextclade dataset get) downloads the latest version of the Nextclade dataset for SARS-CoV-2 (option --name sars-cov-2). We define the directory to store this information with --output-dir.
+
The next step is to run the actual clade assignment on our data. We use the database from the previous step as --input-dataset, we define a directory to output all the results with --output-all and at the end of the command we give as input our clean FASTA consensus genomes.
+
+
More information about running Nextclade from the command line can be found in its online documentation.
+
+
6.3.1 Web Application
+
Nextclade offers an interactive application, which can be used to run its analysis on a FASTA file with sequences:
Click Select a file to browse your computer and upload the FASTA file with the cleaned consensus sequences (results/clean_sequences.fa).
+
Nextclade will automatically detect your data are from SARS-CoV-2, but if not you can select this organism.
+
Click Run.
+
+
Nextclade will show a progress of its analysis at the top of the page, and the results of several quality control metrics in the main panel (see Figure).
+
+
+
+Overview of the Nextclade web interface.
+
+
+
+
+
+
+
+
+Nextclade and data privacy
+
+
+
+
When using the Nextclade web application, the data does not leave your computer, so privacy concerns are not an issue.
+
+
+
+
+
+
6.4 Exercises
+
+
+
+
+
+
+Nextclade
+
+
+
+
+
+
+
+
In this exercise we will work with 48 consensus sequences from the UK, processed with the nf-core/viralrecon pipeline and covered in the previous section.
+
Go to nextclade.org and load the sequences provided in uk_illumina/preprocessed/clean_sequences.fa.
+
+
Are there any samples that were classified as “bad” quality? If so, what is the main reason?
+
Sort the table by the “Clade” column. Looking at the mutations in gene S on the right, you can see that all sequences classified as “Alpha” have a deletion in positions 21992-21994. Samples classified as “Delta” do not have this deletion and instead have a deletion in positions 22029-22034. However, there is one exception: sample GB39, classified as “Delta” has both deletions. Investigate if this mutation is accurate using IGV:
+
+
Open the BAM alignment file for this sample (the alignment file is in results/viralrecon/variants/bowtie2/GB39.ivar_trim.sorted.bam).
+
Open the BAM alignment file for one of the “Alpha” samples as a comparison.
+
Open the ARTIC primer files (two BED files found in resources/primers/).
+
Go to the position where this deletion was identified and investigate if it seems clear from the mapped reads.
+
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
Question 1
+
After loading the samples to the Nextclade web application, we can see that, generally, these samples have high quality as very few are highlighted by Nextclade’s analysis.
+
Sorting the table by the “QC” column, we can see two samples with bad quality (red) and one with mediocre quality (yellow).
+
There are separate reasons for the two bad quality samples:
+
+
Sample GB16 has low quality due to a high number of missing bases.
+
Sample GB39 has low quality due to the occurrence of too many “private mutations”. These are mutations that are only found in this sample, compared to other samples present in Nextclade’s background sequence tree. As we don’t usually expect too many new mutations to occur in a new sample (the mutation rate of SARS-CoV-2 is around 2 new mutations per month), the occurrence of too many private mutations could indicate sequencing quality issues.
+
+
Question 2
+
Sort the table by the “Clade” column. Looking at the mutations in gene S on the right, you can see that all sequences classified as “Alpha” have a deletion in positions 21992-21994. Samples classified as “Delta” do not have this deletion and instead have a deletion in positions 22029-22034. However, there is one exception: sample GB39, classified as “Delta” has both deletions. Investigate if this mutation is accurate using IGV:
+
+
Open the BAM alignment file for this sample (the alignment file is in preprocessed/variants/bowtie2/GB39.ivar_trim.sorted.bam).
+
Open the BAM alignment file for one of the “Alpha” samples as a comparison.
+
Open the ARTIC primer files (two BED files found in resources/primers/).
+
Go to the position where this deletion was identified and investigate if it seems clear from the mapped reads.
+
+
By sorting the table by “Clade”, we can see that although sample GB39 was classified as a Delta variant, it has two deletions in gene S that are present in Alpha variants, as shown in this snapshot (click the image to view a bigger size):
+
+
+
+
As we investigate one of these in more detail from the BAM file (opening it in IGV), we can see that there is an inconsistency between reads coming from the PCR fragment “nCoV-2019_72” and those reads coming from the “nCoV-2019_73” fragment. In some of those reads the deletion is present, but in others it is not. If we look at an Alpha variant sample (for example GB43) we can see that this deletion is present in both cases.
+
+
If we thought this sample was crucial for public health investigation, then this would require further investigation by doing a PCR with new primers and Sanger-sequencing the fragment, for example.
+
+
+
+
+
+
+
+
+
+
+
+
+
6.5 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
Groups of similar SARS-CoV-2 sequences are classified into lineages or clades by different groups. The main nomenclature systems in use are Nextstrain, Pangolin and GISAID.
+
In addition, the World Health Organisation (WHO) classifies some forms of SARS-CoV-2 as variants of concern or variants of interest. These are forms of the virus that have been determined to have a significant public health impact.
+
Both Pangolin and Nextclade assign consensus sequences to lineages/clades and additionally identify those that correspond to WHO variants of concern. Both of these are run as part of the nf-core/viralrecon pipeline, but can also be run using a web application:
+
Besides clade assignment and variant classification, Nextclade provides additional analysis such as identification of mutations and quality control metrics that can be used to identify problematic samples.
Understand the basics of how phylogeny trees are constructed using maximum likelihood methods.
+
Describe what branch lengths represent in typical SARS-CoV-2 phylogenetic tree.
+
Recognise the limitations and challenges in building phylogenies for SARS-CoV-2.
+
Use MAFFT to produce a multiple sequence alignment.
+
Use IQ-Tree for phylogenetic tree inference.
+
Use TimeTree to obtain time-scaled phylogenetic trees using sample collection date metadata.
+
+
+
+
+
+
+
7.1 SARS-CoV-2 Phylogeny
+
+
Building phylogenetic trees for SARS-CoV-2 is challenging due to the large number of sequences available, with millions of genomes submited to GISAID to this day. This means that using maximum-likelihood inference tools to build a global SARS-CoV-2 phylogeny is both time-consuming and computationally demanding.
+
However, several researchers have dedicated their time to identifying tools and models suitable for the phylogenetic analysis of this organism. For example, the global phylogenies repository from Rob Lanfear provide with several tips on building phylogenetic trees for this organism. Their trees are regularly updated and available to download from the GISAID website (which requires an account).
+
Global phylogenies are also available from the groups of Russell Corbett-Detig and Yatish Turakhia, who have developed efficient methods and tools for dealing with large phylogenies. These tools include UShER and matUtils, introducing a new and efficient file format for storing phylogenetic trees, mutations and other annotations (such as lineages) called mutation-annotated trees (MAT format). Their phylogenies are updated daily and are publicly available for download. (Note: Course materials covering these tools are still under development.)
+
Two popular tools used for phylogenetic inference via maximum-likelihood are FastTree and IQ-Tree. Generally, when building a tree from a collection of samples you can include the Wuhan-Hu-1 reference sequence as an outgroup to root the tree. Optionally, you can also add a collection of sequences from around the world (and across time) to contextualise your samples in the global diversity.
+
As an input, these programs need a multiple sequence alignment FASTA file, which is where we start our analysis.
+
+
+
+
+
+
+Data for this section
+
+
+
+
We will work from the course materials folder called 04-phylogeny, which contains the following files:
+
+
data/uk_consensus.fa and data/india_consensus.fa are consensus sequences from the UK and India, respectively. These are the sequences previously assembled using the nf-core/viralrecon pipeline.
+
sample_annotation.tsv is a tab-separated values (TSV) file with information about each sample such as the date of collection and lineage/clade they were assiged to from our analysis. We will use this table to annotate our phylogenetic trees. This table can also be opened in a spreadsheet program such as Excel.
+
resources/reference/sarscov2.fa is the reference genome.
+
+
+
+
+
+
7.2 Alignment
+
The first step in building a phylogenetic tree is to produce a multiple sequence alignment from all our consensus sequences. This is the basis for building a phylogenetic tree from the positions that are variable across samples.
+
A widely used multiple sequence alignment software is called MAFFT. For SARS-CoV-2, a reference-based alignment approach is often used, which is suitable for closely-related genomes. MAFF provides this functionality, which is detailed in its documentation.
+
We demonstrate the analysis using the UK samples in our course materials. First, start by creating a directory for the output:
+
mkdir-p results/mafft
+
Then, we run the command to generate a reference-based alignment:
--6merpair is a fast method for estimating the distances between sequences, based on the number of short 6bp sequences shared between each pair of sequences. This is less accurate than other options available (like --localpair and --globalpair), but runs much faster in whole genome data like we have.
+
--maxambiguous 0.2 automatically removes samples with more than 20% ambiguous ‘N’ bases (or any other value of our choice). This is a convenient way to remove samples with poor genome coverage from our analysis.
+
--addfragments data/consensus_sequences.fa is the FASTA file with the sequences we want to align.
+
finally, at the end of the command we give the reference genome as the input sequence to align our sequences against.
+
+
MAFFT provides several other methods for alignment, with tradeoffs between speed and accuracy. You can look at the full documentation using mafft --man (mafft --help will give a shorter description of the main options). For example, from its documentation we can see that the most precise alignment can be obtained with the options --localpair --maxiterate 1000. However, this is quite slow and may not be feasible for whole genome data of SARS-CoV-2.
+
+
7.2.1 Visualising alignments
+
We can visualise our alignment using the software AliView, which is both lightweight and fast, making it ideal for large alignments. Visualising the alignment can be useful for example to identify regions with missing data (more about this below).
+
+
+
+Snapshop of an alignment visualised with AliView. In this case we are looking at the end of the alignment of our sequences, which shows a typical high number of missing (‘N’) bases.
+
+
+
+
+
+
+
+
+Other Alignment Strategies
+
+
+
+
There are other commonly used alignment tools used for SARS-CoV-2 genomes:
+
+
The minimap2 software has been designed for aligning long sequences to a reference genome. It can therefore be used to align each consensus sequence to the Wuhan-Hu-1 genome. This is the tool internally used by Pangolin.
+
Nextclade uses an internal alignment algorithm where each consensus sequence is aligned with the reference genome. The alignment produced from this tool can also be used for phylogenetic inference.
+
+
It is worth mentioning that when doing reference-based alignment, insertions relative to the reference genome are not considered.
+
+
+
+
+
+
7.3 Tree Inference: IQ-Tree
+
IQ-TREE supports many substitution models, including models with rate heterogeneity across sites.
+
Let’s start by creating an output directory for our results:
+
mkdir-p results/iqtree
+
And then run the program with default options (we set --prefix to ensure output files go to the directory we just created and are named “uk”):
Without specifying any options, iqtree2 uses ModelFinder to find the substituion model that maximizes the likelihood of the data, while at the same time taking into account the complexity of each model (using information criteria metrics commonly used to assess statistical models).
+
From the information printed on the console after running the command, we can see that the chosen model for our alignment was “GTR+F+I”, a generalised time reversible (GTR) substitution model. This model requires an estimate of each base frequency in the population of samples, which in this case is estimated by simply counting the frequencies of each base from the alignment (this is indicated by “+F” in the model name). Finally, the model includes rate heterogeneity across sites, allowing for a proportion of invariant sites (indicated by “+I” in the model name). This makes sense, since we know that there are a lot of positions in the genome where there is no variation in our samples.
+
We can look at the output folder (specified with --prefix) where we see several files with the following extension:
+
+
.iqtree - a text file containing a report of the IQ-Tree run, including a representation of the tree in text format.
+
.treefile - the estimated tree in NEWICK format. We can use this file with other programs, such as FigTree, to visualise our tree.
+
.log - the log file containing the messages that were also printed on the screen.
+
.bionj - the initial tree estimated by neighbour joining (NEWICK format).
+
.mldist - the maximum likelihood distances between every pair of sequences.
+
ckp.gz - this is a “checkpoint” file, which IQ-Tree uses to resume a run in case it was interrupted (e.g. if you are estimating very large trees and your job fails half-way through).
+
.model.gz - this is also a “checkpoint” file for the model testing step.
+
+
The main files of interest are the report file (.iqtree) and the NEWICK tree file (.treefile).
+
+
+
+
+
+
+Inference of very large trees
+
+
+
+
Although running IQ-Tree with default options is fine for most applications, there will be some bottlenecks once the number of samples becomes too large. In particular, the ModelFinder step may be very slow and so it’s best to set a model of our choice based on other people’s work. For example, work by Rob Lanfear suggests that models such as “GTR+G” and “GTR+I” are suitable for SARS-CoV-2. We can specify the model used by iqtree2 by adding the option -m GTR+G, for example.
+
For very large trees (over 10k or 100k samples), using an alternative method to place samples in an existing phylogeny may be more adequate. UShER is a popular tool that can be used to this end. It uses a parsimony-based method, which tends to perform well for SARS-CoV-2 phylogenies.
+
+
+
+
+
7.4 Visualising Trees
+
There are many programs that can be used to visualise phylogenetic trees. In this course we will use FigTree, which has a simple graphical user interface.
+
To open the tree, go to File > Open… and browse to the folder with the IQ-Tree output files. Select the file with .treefile extension and click Open. You will be presented with a visual representation of the tree.
+
We can also import a “tab-separated values” (TSV) file with annotations to add to the tree. For example, we can use our results from Pangolin and Nextclade, as well as other metadata to improve our visualisation (we have prepared a TSV with this information combined, which you could do using Excel or another spreadsheet software).
+
+
Go to File > Import annotations… and open the annotation file.
+
On the menu on the left, click Tip Labels and under “Display” choose one of the fields of our metadata table. For example, you can display the lineage assigned by Pangolin (“pango_lineage” column of our annotation table).
+
+
There are many ways to further configure the tree, including highlighting clades in the tree, and change the labels. See the figure below for an example.
+
+
+
+Annotated phylogenetic tree obtained with FigTree. We used the lineage of each sample as our tip labels and aligned the labels on the right (check the tickbox at the top of the left menu called “Align tip labels”). We identified two clades in the tree that corresponded to the Alpha and Delta variants, and used the “Highlight” tool to give them different colours. To do this, change the “Selection Mode” at the top to “Clade”, then select the branch at the base of the clade you want to highlight, and press the “Highlight” button on the top to pick a colour.
+
+
+
+
+
+
7.5 Time-scaled Phylogenies
+
The trees that we build from sequence data are scaled using the mutation rate estimated from the sequence alignments. This is useful if we want to know, for example, on average how many mutations separate different branches of the tree.
+
Another way to scale trees is to use time. For viral genome sequences, we usually have information about their date of collection, and this can be used to scale the phylogeny using the date information. The idea is to rescale the trees such that the x-axis of the tree represents a date rather than number of mutations.
+
Two programs that can be used to time-scale trees using date information are called TreeTime and Chronumental. Chronumental was developed for dealing with very large phylogenies (millions of samples), but lacks some of the functionalities provided by TreeTime (such as estimating uncertainty in date estimates, reconstructing past sequences, re-rooting trees, among others). So, if you are working with less than ~50,000 sequences, we recommend using TreeTime, otherwise Chronumental is a suitable alternative.
+
Because we are dealing with a small number of samples, we will show an example of using TreeTime to scale our tree based on our dates:
After running, this tool produces several output files in the specified folder. The main files of interest are:
+
+
timetree.nexus is the new date-scaled tree. NEXUS is another tree file format, which can store more information about the tree compared to the simpler NEWICK format. This format is also supported by FigTree.
+
timetree.pdf is a PDF file with the inferred tree, including a time scale on the x-axis. This can be useful for quick visualisation of the tree.
+
+
We can visualise this tree in FigTree, by opening the file timetree.nexus. The scale of this new tree now corresponds to years (instead of nucleotide substitution rates). We make make several adjustments to this tree, to make it look how we prefer. For example:
+
+
Label the nodes of the tree with the inferred dates: from the left menu click Node Labels and under “Display” select “date”.
+
Import the metadata table (sample_annotation.tsv file) and display the results of Nextclade or Pangolin clades/lineages.
+
Adjust the scale of the tree to be more intuitive. For example, instead of having the unit of the scale in years, we can change it to months. On the left menu, click Time Scale and change “Scale factor” to 12 (twelve months in a year). Then click Scale Bar and change the “Scale range” to 1. The scale now represents 1 month of time, which may be easier to interpret in this case.
+
+
Note that there is uncertainty in the estimates of the internal node dates from TimeTree and these should be interpreted with some caution. The inference of the internal nodes will be better the more samples we have, and across a wider range of times. In our specific case we had samples all from a similar period of time, which makes our dating of internal nodes a little poorer than might be desired.
+
More precise tree dating may be achieved by using public sequences across a range of times or by collecting more samples over time. Time-scaled trees of this sort can therefore be useful to infer if there is a recent spread of a new clade in the population.
+
+
+
7.6 Missing Data & Problematic Sites
+
So far, we have been using all of our assembled samples in the phylogenetic analysis. However, we know that some of these have poorer quality (for example the “IN01” sample from India had low genome coverage). Although, generally speaking, sequences with missing data are unlikely to substantially affect the phylogenetic results, their placement in the phylogeny will be more uncertain (since several variable sites may be missing data). Therefore, for phylogenetic analysis, it is best if we remove samples with low sequencing coverage, and instead focus on high-quality samples (e.g. with >80% coverage).
+
A more serious issue affecting phylogenies is the presence of recurrent errors in certain positions of the genome. One of the regions with a higher prevalence of errors is the start and end of the consensus sequence, which also typically contains many missing data (see example in Figure 2). Therefore, it is common to mask the first and last few bases of the alignment, to avoid including spurious variable sites in the analysis.
+
+
+
+
+
+
+Sequence masking
+
+
+
+
The term masking is often used to refer to the process of converting sequence bases to the ambiguous character ‘N’. You may come across this term in the documentation of certain tools, for example: “Positions with less than 20x depth of sequencing are masked.”
+
Masks are not limited to depth of sequencing. For example, reference genomes from ENSEMBL are available with masked repeat or low-complexity sequences (e.g. around centromeres, transposon-rich regions, etc.).
+
The term soft masking is also used to refer to cases where, instead of using the ambiguous character ‘N’, sequences are masked with a lowercase. For example:
Additionally, work by Turakhia, de Maio, Thornlow, et al. (2020) has identified several sites that show an unexpected mutation pattern. This includes, for example, mutations that unexpectedly occur multiple times in different parts of the tree (homoplasies) and often coincide with primer binding sites (from amplicon-based protocols) and can even be lab-specific (e.g. due to their protocols and data processing pipelines). The work from this team has led to the creation of a list of problematic sites, which are recommended to be masked before running the phylogenetic analysis.
So, let’s try to improve our alignment by masking the problematic sites, which are provided as a VCF file. This file also includes the first and last positions of the genome as targets for masking (positions 1–55 and 29804–29903, relative to the Wuhan-Hu-1 reference genome MN908947.3). The authors also provide a python script for masking a multiple sequence alignment. We have already downloaded these files to our course materials folder, so we can go ahead and use the script to mask our file:
If we open the output file with AliView, we can confirm that the positions specified in the VCF file have now been masked with the missing ‘N’ character.
+
We could then use this masked alignment for our tree-inference, just as we did before.
+
+
+
+
+
+
+Using Python Scripts
+
+
+
+
Bioinformaticians often write custom scripts for particular tasks. In this example, the authors of the “problematic sites” wrote a Python script that takes as input the FASTA file we want to mask as well as a VCF with the list of sites to be masked.
+
Python scripts are usually run with the python program and often accept options in a similar way to other command-line tools, using the syntax --option (this is not always the case, but most professionally written scripts follow this convention). To see how to use the script we can use the option --help. For our case, we could run:
+
python scripts/mask_alignment_using_vcf.py --help
+
+
+
+
+
7.7 Exercises
+
+
+
+
+
+
+Building phylogenies
+
+
+
+
+
+
+
+
So far we have focused our analysis on the samples from the UK. In this exercise you will be able to practice these steps on the samples from India. The steps are similar to what we have done so far, and you can consult the materials in the sections above to go through each exercise.
+
In the following exercises, you can run the commands directly from the command line. But if you feel comfortable with nano, as a bonus, you can try to save the commands in a shell script.
+
+
Using mafft, produce a multiple-sequence alignment from the India consensus sequences in the file data/india_consensus.fa. Save the output in a file named results/mafft/india_alignment.fa.
+
Using iqtree2, infer a phylogenetic tree from this alignment. Save the output with prefix results/iqtree/india.
+
+
What substitution model did IQ-Tree infer as the most likely for the data?
+
+
Using FigTree, visualise the inferred tree:
+
+
Open the .tree output file.
+
Import the annotation table sample_annotation.tsv.
+
Make the tip labels display the Nextclade clade instead of the sample names.
+
Highlight any clusters of the tree containing WHO variants of concern.
+
+
Using timetree, re-scale the phylogeny using dates (the file sample_annotation.tsv can be used as input to timetree along with the previously-created phylogeny and alignments). Output the result to results/treetime/india
+
+
Open the .nexus output file in FigTree.
+
Make the node labels display the date inferred by timetree.
+
+
Two samples in the time-scaled tree appear at the root of the tree: IN05 and IN33. But we would have expected the reference sample (MN908947.3) to be the root of the tree, as it was collected in Dec 2019.
+
+
Investigate what lineages these samples were assigned to.
+
Go to https://cov-lineages.org/lineage_list.html and check when those lineages were detected.
+
Is the collection date metadata for these samples compatible with the lineage information from the Pangolin website? Can you hypothesise what may have happened with these samples?
This command prints a lot of output on the screen. We can see all this information (and more!) on the output file results/iqtree/india.iqtree, which contains a log of all the analysis done by the program. We could, for example, use the less program to look inside this file. When we do this, we can see the following:
+
ModelFinder
+-----------
+
+Best-fit model according to BIC: GTR+F+I
+
Which indicates that the substitution model used by IQ-Tree was “GTR+F+I”. This is the same model that was determined for the UK samples, and an explanation of this model is given in the materials above.
+
Question 3
+
Using FigTree:
+
+
Go to File > Open… and browse to the folder with the IQ-Tree output files.
+
Select the file with india.treefile and click Open.
+
Go to File > Import annotations… and open the annotation file sample_annotation.tsv.
+
On the menu on the left, click Tip Labels and under “Display” choose the field “nextclade_clade”.
+
+
On this tree, there is a small group of samples classified as “20I (Alpha; V1)”. These samples correspond to the Alpha variant of concern. To highlight these:
+
+
Change the “Selection Mode” at the top to “Clade”.
+
Select the branch corresponding to the base of the group of samples classified as Alpha. This should highlight all those branches.
+
Click the “Highlight” button at the top and choose a colour.
Once complete, we can open the india.nexus tree with FigTree. We can annotate the internal nodes of the tree with the dates inferred by treetime by clicking on the Node Labels menu on the left and selecting “Display” to be “date”.
+
+
Question 5
+
To investigate this strange result, we open the sample_annotation.tsv file in our spreadsheet program. We can see the following information for these two samples:
+
name date country year month pango_lineage nextclade_clade
+IN05 2021-01-21 India 2021 1 A.23.1 19B
+IN33 2021-01-21 India 2021 1 A.23.1 19B
+
Both these samples were assigned to Pango lineage “A.23.1”. From the cov-lineages.org website we can see that these were detected as early as 2020-06-08.
+
However, our samples’ metadata indicate that these samples were collected in January 2021, that is 7 months after these samples were first detected globally.
+
This contractiction explains the strange result in our time-scaled tree. These samples will have mutations that are part of the older lineage “A”, but were annotated to be from 2021. So treetime put them down at the root of the tree.
+
This discrepancy could indicate an issue with the metadata collection, which perhaps is wrong. For example, it could be that these samples were collected months before January 2021, but only sequenced then. And by mistake the date of sequencing was recorded as the date of collection.
+
This is an example how important accurate metadata is for our analysis.
+
+
+
+
+
+
+
+
+
+
+
+
+
7.8 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
Methods for phylogenetic inference include parsimony and maximum likelihood. Maximum likelihood methods are preferred because they include more features of the evolutionary process. However, they are computationally more demanding than parsimony-based methods.
+
To build a phylogenetic tree we need a multiple sequence alignment of the sequences we want to infer a tree from.
+
In SARS-CoV-2, alignments are usually done against the Wuhan-Hu-1 reference genome.
+
We can use the software mafft to produce a multiple sequence alignment. The option --addfragments is used to produce an alignment against the reference genome.
+
The software iqtree2 can be used for inferring trees from an alignment using maximum likelihood. This software supports a wide range of substitution models and a method to identify the model that maximizes the likelihood of the data.
+
Some of the substituion models that have been used to build global SARS-CoV-2 phylogenies are “GTR+G” and “GTR+I”.
+
We can time-scale trees using sample collection date information. The program treetime can be used to achieve this. For very large sample sizes (>50,000 samples) the much faster program Chronumental can be used instead.
+
Before building a phylogeny, we should be careful to mask problematic sites that can lead to misleading placements of samples in the tree. The SARS-CoV-2 Problematic Sites repository provides with an updated list of sites that should be masked.
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
\ No newline at end of file
diff --git a/materials/02-isolates/images/alignment_aliview.png b/materials/02-isolates/images/alignment_aliview.png
new file mode 100644
index 0000000..5b2848e
Binary files /dev/null and b/materials/02-isolates/images/alignment_aliview.png differ
diff --git a/materials/02-isolates/images/igv_amplicon_dropout.svg b/materials/02-isolates/images/igv_amplicon_dropout.svg
new file mode 100644
index 0000000..f831e87
--- /dev/null
+++ b/materials/02-isolates/images/igv_amplicon_dropout.svg
@@ -0,0 +1,281 @@
+
+
diff --git a/materials/02-isolates/images/igv_deletion_example.svg b/materials/02-isolates/images/igv_deletion_example.svg
new file mode 100644
index 0000000..520ec6d
--- /dev/null
+++ b/materials/02-isolates/images/igv_deletion_example.svg
@@ -0,0 +1,469 @@
+
+
+
+
diff --git a/materials/02-isolates/images/lineages_example.svg b/materials/02-isolates/images/lineages_example.svg
new file mode 100644
index 0000000..c503d4c
--- /dev/null
+++ b/materials/02-isolates/images/lineages_example.svg
@@ -0,0 +1,4282 @@
+
+
+
+
diff --git a/materials/02-isolates/images/nextclade_deletion_example.svg b/materials/02-isolates/images/nextclade_deletion_example.svg
new file mode 100644
index 0000000..b609f77
--- /dev/null
+++ b/materials/02-isolates/images/nextclade_deletion_example.svg
@@ -0,0 +1,3770 @@
+
+
+
+
diff --git a/materials/02-isolates/images/nextclade_overview.svg b/materials/02-isolates/images/nextclade_overview.svg
new file mode 100644
index 0000000..4878e97
--- /dev/null
+++ b/materials/02-isolates/images/nextclade_overview.svg
@@ -0,0 +1,2919 @@
+
+
+
+
diff --git a/materials/02-isolates/images/phylogeny_figtree.svg b/materials/02-isolates/images/phylogeny_figtree.svg
new file mode 100644
index 0000000..037eeef
--- /dev/null
+++ b/materials/02-isolates/images/phylogeny_figtree.svg
@@ -0,0 +1,3018 @@
+
+
diff --git a/materials/02-isolates/images/switzerland_variants.svg b/materials/02-isolates/images/switzerland_variants.svg
new file mode 100644
index 0000000..346e5d0
--- /dev/null
+++ b/materials/02-isolates/images/switzerland_variants.svg
@@ -0,0 +1,2141 @@
+
+
+
+
diff --git a/materials/02-isolates/images/variants_snps_indels.svg b/materials/02-isolates/images/variants_snps_indels.svg
new file mode 100644
index 0000000..f1ff382
--- /dev/null
+++ b/materials/02-isolates/images/variants_snps_indels.svg
@@ -0,0 +1,376 @@
+
+
+
+
diff --git a/materials/02-isolates/images/viralrecon_coverage_heatmap.png b/materials/02-isolates/images/viralrecon_coverage_heatmap.png
new file mode 100644
index 0000000..93cd1d4
Binary files /dev/null and b/materials/02-isolates/images/viralrecon_coverage_heatmap.png differ
diff --git a/materials/02-isolates/images/viralrecon_multiqc_plot.png b/materials/02-isolates/images/viralrecon_multiqc_plot.png
new file mode 100644
index 0000000..a1e9fba
Binary files /dev/null and b/materials/02-isolates/images/viralrecon_multiqc_plot.png differ
diff --git a/materials/02-isolates/images/viralrecon_multiqc_variant_metrics.svg b/materials/02-isolates/images/viralrecon_multiqc_variant_metrics.svg
new file mode 100644
index 0000000..3f459c0
--- /dev/null
+++ b/materials/02-isolates/images/viralrecon_multiqc_variant_metrics.svg
@@ -0,0 +1,4304 @@
+
+
diff --git a/materials/02-isolates/images/viralrecon_pipeline_info_duration.png b/materials/02-isolates/images/viralrecon_pipeline_info_duration.png
new file mode 100644
index 0000000..f910a99
Binary files /dev/null and b/materials/02-isolates/images/viralrecon_pipeline_info_duration.png differ
diff --git a/materials/02-isolates/images/viralrecon_workflow_illumina.svg b/materials/02-isolates/images/viralrecon_workflow_illumina.svg
new file mode 100644
index 0000000..8a202c5
--- /dev/null
+++ b/materials/02-isolates/images/viralrecon_workflow_illumina.svg
@@ -0,0 +1,1341 @@
+
+
diff --git a/materials/02-isolates/images/viralrecon_workflow_medaka.svg b/materials/02-isolates/images/viralrecon_workflow_medaka.svg
new file mode 100644
index 0000000..5813c2f
--- /dev/null
+++ b/materials/02-isolates/images/viralrecon_workflow_medaka.svg
@@ -0,0 +1,1286 @@
+
+
diff --git a/materials/02-isolates/images/workflow_overview.svg b/materials/02-isolates/images/workflow_overview.svg
new file mode 100644
index 0000000..0adb444
--- /dev/null
+++ b/materials/02-isolates/images/workflow_overview.svg
@@ -0,0 +1,1721 @@
+
+
diff --git a/materials/03-case_studies/01-switzerland.html b/materials/03-case_studies/01-switzerland.html
new file mode 100644
index 0000000..637396c
--- /dev/null
+++ b/materials/03-case_studies/01-switzerland.html
@@ -0,0 +1,1173 @@
+
+
+
+
+
+
+
+
+
+SARS Genomic Surveillance
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
8Switzerland (Nanopore)
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Learning Objectives
+
+
+
+
This section demonstrates a start-to-finish analysis of a dataset sequenced on a Nanopore platform, using the concepts and tools covered in previous sections. You can download the data from these links (two versions available):
+
+
Switzerland Case Study - Full Data – this includes data for 65 samples, which gives a more realistic sample size, but can take several hours to run on a small computer.
+
Switzerland Case Study - Small Version – this includes data for a subset of 10 samples, which is more suitable for training purposes (but the results will look slightly different from the ones shown here).
+
+
By the end of this section, you should be able to:
+
+
Prepare all the files necessary to run the consensus pipeline.
+
Run the viralrecon pipeline to generate FASTA consensus from raw FASTQ files.
+
Assess and collect several quality metrics for the consensus sequences.
+
Clean output files, in preparation for other downstream analysis.
+
Assign sequences to lineages using Nextclade and/or Pangolin.
+
Contextualise your sequences in other background data and cluster them based on phylogenetic analysis.
+
Integrate the metadata and results to generate useful visualisations of your data.
+
Report your analysis.
+
+
+
+
We will analyse data from 48 samples collected in Switzerland between Nov 2021 and Jan 2022. The samples were sequenced on a GridION platform using pore version 9.4.1.
+
The final product of our work (and main objective) is to produce a report of the analysis, which you can see here: Switzerland Case Study Report.
+
In summary, the report addresses the following:
+
+
What was the quality of the consensus sequences?
+
What lineage/clade was each of our samples assigned to?
+
How many clusters of samples did we identify?
+
How did the detected lineages change over the time of sampling?
+
+
We also produce several essential output files, which would usually be necessary to upload our data to public repositories:
+
+
Metadata (CSV)
+
Consensus sequences (FASTA)
+
Consensus sequence quality metrics (CSV)
+
Variants (CSV)
+
+
+
8.1 Pipeline Overview
+
Our analysis starts with FASTQ files, which will be used with the nf-core/viralreconNextflow pipeline. This will give us several quality control metrics essential for our downstream analysis and reporting.
+
Critical files output by the pipeline will need to be further processed, including combining our consensus FASTA files and obtaining a list of filtered SNP/indel variants. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of mutations or variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions.
+
+
+
+
8.2 Preparing Files
+
Before we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. From the data we are starting with, we already have the following directories:
+
+
data → contains the sequencing data in a sub-directory called fast_pass.
+
resources → files that were downloaded from public repositories.
+
scripts → bash and R scripts used to run the analysis.
+
+
We create two additional directories:
+
+
report → files and documents that we report to our colleagues or upload to public repositories.
+
results → results of the analysis.
+
+
You can create directories from the command line using the mkdir command:
+
mkdir results
+mkdir report
+
+
8.2.1 Data
+
We start our analysis from FASTQ files generated using the software Guppy v6.1.5 ran in “fast” mode. This software outputs the files to a directory called fastq_pass, with further sub-directories for each sample barcode. This is how it looks like in this case:
Metadata for these samples is available in the file sample_info.csv. Here is some of the information we have available for these samples:
+
+
sample → the sample ID.
+
collection_date → the date of collection for the sample in the format YYYY-MM-DD.
+
country → the country of origin for this sample.
+
latitude/longitude → coordinates for sample location (optional).
+
sequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).
+
sequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).
+
amplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)
+
Specific columns for Oxford Nanopore data, which are essential for the bioinformatic analysis:
+
+
ont_pore → the version of the pores.
+
ont_guppy_version → the version of the Guppy software used for basecalling.
+
ont_guppy_mode → the basecalling mode used with Guppy.
+
+
+
+
+
+
8.3 Consensus Assembly
+
+
+
+
+
+
+Note
+
+
+
+
See Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.
+
+
+
The first step in the bioinformatic analysis is to run the nf-core/viralrecon pipeline.
+
+
8.3.1 Samplesheet
+
But first we need to prepare our input files. For Nanopore data, we need a samplesheet CSV file with two columns, indicating sample name (first column) and the respective barcode number (second column).
+
We produced this table in Excel and saved it as a CSV file. Here are the top few rows of the file:
Now we are ready to run the nf-core/viralrecon pipeline (see Section 4.3 for details). We saved our command in a script (scripts/01-run_viralrecon.sh), which we created with the command line text editor nano. This ensures that our analysis is reproducible and traceable (we can go back to the script to see how the analysis was run).
+
First, we activate our software environment, to ensure Nextflow is available to us:
+
mamba activate nextflow
+
Then, we run our script with:
+
bash scripts/01-run_viralrecon.sh
+
Which will start executing the pipeline.
+
For reference, here is the command included in that script:
In this case, we used the medaka model r941_min_fast_g303, because that is the latest one available (even though our Guppy version is 6.1.5). We also restricted our --max_memory and --max_cpus due to the size of our processing computers. If a larger computer was available, we could have used higher values for these parameters.
+
+
+
+
+
+
+The \ in long commands
+
+
+
+
In the command above, you will notice several \ at the end of each line. This is indicating that we want to continue writing our command in the next line. Notice that the last line does not include \, because that is the end of the command. This is very useful when commands are very long, because it makes the code more readable.
+
+
+
+
+
+
8.4 Consensus Quality
+
+
+
+
+
+
+Note
+
+
+
+
See Section 5.2, if you need to revise how to assess the quality of consensus sequences.
+
+
+
+
8.4.1 General Metrics
+
We used the MultiqQC report to assess the initial quality of our samples. The quality report can be found in results/viralrecon/multiqc/medaka/multiqc_report.html.
+
We paid particular attention to:
+
+
Number of reads mapped to the reference genome.
+
Median depth of coverage.
+
Percentage of the genome with missing bases (‘N’).
+
Number of SNP + Indel variants.
+
+
+
+
+Example MultiQC output, highlighting the main columns with quality information that will be collected for the final report.
+
+
+
We noted that:
+
+
9 samples had more than 15% missing bases.
+
All samples had median depth of coverage greater than 20x.
+
There was some systematic dropout for some amplicons, in particular nCoV-2019_64 had very low amplification in several of the samples. Of note was also nCoV-2019_73, and other neighbouring amplicons.
+
+
+
+
+Amplicon depth of sequencing (or coverage) across samples, from the MultiQC report. Darker regions indicate systematic amplicon dropout, probably due to issues during the PCR amplification.
+
+
+
Besides the MultiQC report, the pipeline also outputs a CSV file with collected summary metrics (equivalent to the first table on the report): results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv. We will use this file later to join this information with our metadata and lineage assignment using the R software (detailed in “Integration & Visualisation” section, below).
+
+
+
8.4.2 Variants
+
We also looked at the table of variants obtained from the pipeline. This is output in results/viralrecon/medaka/variants_long_table.csv. This table can be very useful to keep track of particular mutations that may be increasing over time. Later, we will tidy this table to attach to our reported results (“Integration & Visualisation” section).
+
But for now, we will explore this table to address a few more quality-related questions. We opened this table in Excel to answer the following:
+
+
Where there samples with a high number of intermediate allele frequencies? This could indicate mixed samples due to cross-contamination.
+
Where there samples with frameshift mutations? These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.
+
+
All the variants in our samples had an alternative allele frequency (AF column) greater than 68%, which is a good indication that there were no mixed samples. We did note two samples – CH13 and CH49 – had a frameshift mutation each, in the Spike and ORF1a proteins, respectively. This may be due to a sequencing error, as it is not observed in any other samples. The inclusion of this error may not affect our downstream analysis significantly, but we make a note of these two samples, to check if their lineage assignment and phylogenetic analysis makes sense later on (or whether they appear as outliers, indicating broader sequencing quality issues).
+
+
+
+Variants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases.
+
+
+
+
+
8.4.3 Clean FASTA
+
The pipeline outputs the consensus sequences in results/viralrecon/medaka/*.consensus.fasta (one file for each sample). For downstream analysis, it is convenient to combine all these sequences into a single file, and also clean the sequence names (to remove some text – “/ARTIC/medaka MN908947.3” –, which is added by the medaka variant caller).
+
We created a new script to clean our consensus FASTA files, which we ran with bash scripts/02-clean_fasta.sh:
+
#!/bin/bash
+
+# combine and clean FASTA files
+cat results/viralrecon/medaka/*.consensus.fasta |sed's|/ARTIC/medaka MN908947.3||'> report/consensus.fa
+
This command does two things:
+
+
Combine all our FASTA consensus sequences into a single file (using cat).
+
Clean the sequence names (using sed).
+
+
The output was saved as a new FASTA file: report/consensus.fa.
+
+
+
8.4.4 Missing Intervals
+
As a further quality check, we also generated a table of missing intervals (indicated by the N character in the FASTA sequences). We used the seqkit software to achieve this.
+
First, we activate our software environment:
+
mamba activate seqkit
+
Then, we ran the script bash scripts/03-missing_intervals.sh, which includes the following command:
This software outputs a tab-delimited table, which we saved as results/missing_intervals.tsv. The table looks like this (only the top few rows are shown):
We opened this file missing_intervals.tsv in Excel and quickly calculated the length of each interval. We noted that two samples – CH07 and CH59 – both have a continuous interval of 5130 missing bases between positions 19580 and 24709. This includes part of the ORF1a gene and nearly all of the S (Spike) gene. This may be due to a poor amplification of one of the PCR amplicons and may affect the interpretation of the results for these two samples. We make a note of these samples as being possibly problematic in downstream analysis steps.
+
+
+
+
8.5 Downstream Analyses
+
Based on the clean consensus sequences, we then perform several downstream analysis.
+
+
8.5.1 Lineage Assignment
+
+
+
+
+
+
+Note
+
+
+
+
See Section 6.1, if you need to revise how lineage assignment works.
+
+
+
Although the Viralrecon pipeline runs Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). An up-to-date run of both of these tools can be done using each of their web applications:
However, for automation, reproducibility and traceability purposes, we used the command line versions of these tools, and included their analysis in two scripts.
For Nextclade, we first activate the software environment:
+
mamba activate nextclade
+
And then we ran the script bash scripts/04-nextclade.sh, which contains the following commands:
+
# get nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa
+
The first command downloads the latest version of the Nextclade background data using nextclade dataset. We use that data as input to the second command (nextclade run) to make sure it runs with the most up-to-date lineages.
+
+
+
For Pangolin, we first activate the software environment:
+
mamba activate pangolin
+
And then we ran the script bash/04-pangolin.sh, which contains the following commands:
+
# update pangolin data
+pangolin--update-data
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa
+
Similarly to before, we first ran pangolin --update-data to ensure we were using the latest lineages available. We can check the version of the data used with pangolin --all-versions (at the time we ran this we had pangolin-data: 1.23.1).
+
+
+
+
Both of these tools output CSV files, which can be open in Excel for further examination.
+Opening the pangolin results (results/pangolin/pango_report.csv), we noticed that two samples – CH07 and CH59 – failed the QC due to high fraction of missing data. These are the same two samples that had a large gap of missing data in the previous section. Several other samples were classified as “Probable Omicron”, which from the “scorpio_notes” column we can see may be because too many of the expected mutations had missing (ambiguous) bases in those consensus sequences.
+Opening the nextclade results (results/nextclade/nextclade.tsv), we noticed that 23 samples were assigned a QC status of “bad”, mostly due to high percentage of missing data (nextclade uses a stringent threshold of 3000 sites, or ~10%, missing data).
+
Like before, we will do further analysis (and visualisation) of these data using the software R, in the section “Integration & Visualisation”, detailed below.
+
+
+
8.5.2 Phylogeny
+
+
+
+
+
+
+Note
+
+
+
+
See Section 7.1, if you need to revise how to build phylogenetic trees.
+
+
+
Although a tool such as Nextclade can place our samples in a global phylogeny context, sometimes it may be convient to build our own phylogenies. This requires three steps:
+
+
Producing a multiple sequence alignment from all consensus sequences.
+
Tree inference.
+
Tree visualisation and annotation.
+
+
Before our analysis, we first activated our software environment:
+
mamba activate phylo
+
We performed the first two steps with the following script, which we ran with bash scripts/05-phylogeny.sh:
The output of iqtree includes a tree file, which can be visualised using FigTree (or online using Microreact). The figure below shows an example of an annotated tree, where we highlight the main VOCs detected. This annotation was done based on the file that we generate in the next section (“Integration & Visualisation”), so those steps would have to be done first.
+
We can see that our samples fall broadly into two large clusters, which correlate with the VOC classification for these samples.
+
+
+
+Phylogenetic tree for our samples. The top cluster (purple) highlights samples classified as Omicron (BA.1-related) and the bottom cluster (pink) samples classified as Delta (AY lineage, also known as B.1.617.2). The tips of the tree are coloured according to Nextclade’s QC status: green = “good”; blue = “mediocre”; red = “bad”.
+
+
+
+
+
8.5.3 Clustering
+
We identified groups of similar sequences in our data using the software civet (Cluster Investigation and Virus Epidemiology Tool). This software compares our samples with a background dataset of our choice, which givus us more context for our analysis. In this case we are using the example background data that comes with civet. However, in a real-world analysis, it would have been ideal to choose local samples as background data. For example, we could download samples from Switzerland from around the time period of our sample collection, from GISAID following the instructions on the civet documentation (you need an account on GISAID to obtain these data).
+
For this example, we already prepared civet background dataset saved in resources/civet_background_data.
+
Before our analysis, we first activate our software environment:
+
mamba activate civet
+
Then, we ran the script bash scripts/06-civet.sh, which contains the following code:
The result of this analysis includes an interactive HTML report (in results/civet/civet.html). We can see that our samples were grouped into 2 catchments, using this background data. This makes sense from our previous lineage/variant analysis: the two catchments correlate with the two main variants in these data (Omicron and Delta).
+
+
+
+Example of results from civet report for catchment 2, showing the phylogeny in the context of the samples in the background data (coloured according to their lineages).
+
+
+
Civet also outputs a CSV file (results/civet/master_metadata.csv), which includes the catchment that each sample was assigned to. We will use this CSV file later to integrate this information with other parts of our analysis, in R, detailed in the “Integration & Visualisation” section.
+
+
+
+
8.6 Integration & Visualisation
+
At this point in our analysis, we have several tables with different pieces of information:
+
+
sample_info.csv → the original table with metadata for our samples.
+
results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.
+
results/nextclade/nextclade.tsv → the results from Nextclade.
+
results/pangolin/pango_report.csv → the results from Pangolin.
+
results/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.
+
+
To consolidate our analysis, we tidied and integrated the information from across these different files, into a single table using the software R. The script used to do this is in scripts/07-data_integration.R. Because this is an R script, we opened it in RStudio to execute the code.
+
The output of our script is a new tab-delimited table, which we saved in report/consensus_metrics.tsv, and contains the following columns:
+
+
sample → sample ID.
+
collection_date → date of collection day.
+
collection_week → date of collection week (useful for summarising/visualising counts per-week).
+
country → country of origin.
+
latitude/longitude → latitude and longitude of collection.
+
n_mapped_reads → number of mapped reads.
+
median_depth → median depth.
+
pct_missing → percentage of missing data.
+
pct_coverage → percentage of coverage.
+
n_variants → number of SNP + indel variants detected.
+
nextclade → nextclade clade.
+
qc_status → QC status as determined by Nextclade (“bad”, “mediocre”, “good”).
+
lineage → Pangolin lineage.
+
who_variant → variant of concern designation.
+
catchment → catchment group from Civet.
+
+
This table, which aggregates information from many of the tools we used, was then used to produce different visualisations of our analysis. These visualisations were also done using the R software (scripts/08-visualisation.R), and integrated into a report, shown below.
+
+
+
+
8.7 Bonus: Full Workflow
+
Although we have ran each of the steps of our analysis individually (each in their own script), now that we have everything working, we could integrate all these steps into a single “master” script:
+
#!/bin/bash
+
+# make mamba activate command available
+eval"$(conda shell.bash hook)"
+source$(mamba info --base)/etc/profile.d/mamba.sh
+
+# make report directory
+mkdir-p report
+
+mamba activate nextflow
+
+# run viralrecon
+nextflow run nf-core/viralrecon \
+-r 2.6.0 -profile singularity \
+--max_memory'16.GB'--max_cpus 8 \
+--platform nanopore \
+--input samplesheet.csv \
+--fastq_dir data/fastq_pass/ \
+--outdir results/viralrecon \
+--protocol amplicon \
+--genome'MN908947.3'\
+--primer_set artic \
+--primer_set_version 3 \
+--artic_minion_caller medaka \
+--artic_minion_medaka_model r941_min_fast_g303 \
+--skip_assembly--skip_asciigenome\
+--skip_pangolin--skip_nextclade
+
+# combine and clean FASTA files
+cat results/viralrecon/medaka/*.consensus.fasta |sed's|/ARTIC/medaka MN908947.3||'> report/consensus.fa
+
+mamba activate seqkit
+
+# create missing bases TSV file
+seqkit locate -i-P-G-M-r-p"N+" report/consensus.fa > results/missing_intervals.tsv
+
+mamba activate nextclade
+
+# get nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa
+
+mamba activate pangolin
+
+# update pangolin data
+pangolin--update-data
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa
+
+mamba activate phylo
+
+# alignment
+mkdir-p results/mafft
+mafft--6merpair--maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa
+
+# tree inference
+mkdir-p results/iqtree
+iqtree2-s results/mafft/alignment.fa --prefix results/iqtree/consensus
+
+# data integration and cleaning
+Rscript scripts/07-data_integration.R
+
Notice that we included the R script that does the data cleaning here, using the Rscript program that allows to execute an R script from the command-line.
+
Having this “master” script, we could run all these steps from start-to-finish with a single command, which can be very useful if you want to fully automate your analysis across multiple runs.
This section demonstrates a start-to-finish analysis of a dataset sequenced on an Illumina platform, using the concepts and tools covered in previous sections. You can download the data from these links (two versions available):
+
+
South Africa Case Study - Full Data – this includes data for 24 samples, which gives a more realistic sample size, but can take a few hours to run on a small computer.
+
South Africa Case Study - Small Version – this includes data for a subset of 8 samples, which is more suitable for training purposes (but the results will look slightly different from the ones shown here).
+
+
By the end of this section, you should be able to:
+
+
Prepare all the files necessary to run the consensus pipeline.
+
Run the viralrecon pipeline to generate FASTA consensus from raw FASTQ files.
+
Assess and collect several quality metrics for the consensus sequences.
+
Clean output files, in preparation for other downstream analysis.
+
Assign sequences to lineages using Nextclade and/or Pangolin.
+
Contextualise your sequences in other background data and cluster them based on phylogenetic analysis.
+
Integrate the metadata and results to generate useful visualisations of your data.
+
Report your analysis.
+
+
+
+
We will analyse data from 24 samples collected in South Africa between Nov-Dec 2021. The samples were sequenced on an Illumina MiSeq platform.
+
The final product of our work (and main objective) is to produce a report of the analysis, which you can see here: South Africa Case Study Report.
+
In summary, the report addresses the following:
+
+
What was the quality of the consensus sequences?
+
What lineage/clade was each of our samples assigned to?
+
How many clusters of samples did we identify?
+
How did the detected lineages change over the time of sampling?
+
+
We also produce several essential output files, which would usually be necessary to upload our data to public repositories:
+
+
Metadata (CSV)
+
Consensus sequences (FASTA)
+
Consensus sequence quality metrics (CSV)
+
Variants (CSV)
+
+
+
9.1 Pipeline Overview
+
Our analysis starts with FASTQ files, which will be used with the nf-core/viralreconNextflow pipeline. This will give us several quality control metrics essential for our downstream analysis and reporting.
+
Critical files output by the pipeline will need to be further processed, including combining our consensus FASTA files and obtaining a list of filtered SNP/indel variants. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of mutations or variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions.
+
+
+
+
9.2 Preparing Files
+
Before we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. From the data we are starting with, we already have the following directories:
+
+
data → contains the sequencing data in a sub-directory called reads.
+
resources → files that were downloaded from public repositories.
+
scripts → bash and R scripts used to run the analysis.
+
+
We create two additional directories:
+
+
report → files and documents that we report to our colleagues or upload to public repositories.
+
results → results of the analysis.
+
+
You can create directories from the command line using the mkdir command:
+
mkdir results
+mkdir report
+
+
9.2.1 Data
+
We start our analysis from FASTQ files generated by the Illumina sequencer. As this is paired-end sequencing, we have two files per sample (with suffix _1 and _2):
Metadata for these samples is available in the file sample_info.csv. Here is some of the information we have available for these samples:
+
+
sample → the sample ID.
+
collection_date → the date of collection for the sample in the format YYYY-MM-DD.
+
country → the country of origin for this sample.
+
geo_loc_region → the region within the country where the sample was collected.
+
latitude/longitude → coordinates for sample location (in this case we’re only given a single coordinate for the whole country - in a real setting you may want to collect a precise location).
+
sequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).
+
sequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).
+
amplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)
+
+
+
+
+
9.3 Consensus Assembly
+
+
+
+
+
+
+Note
+
+
+
+
See Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.
+
+
+
The first step in the bioinformatic analysis is to run the nf-core/viralrecon pipeline. But first we need to prepare our input files.
+
+
9.3.1 Samplesheet
+
For Illumina data, we need a samplesheet CSV file with three columns, indicating sample name (first column) and the respective FASTQ file paths for read 1 (second column) and read 2 (third column).
+
Because our FASTQ file names are not very user-friendly, we used some command-line tricks to help us produce this table:
+
# list read 1 files and save output in a temporary file
+ls data/reads/*_1.fastq.gz > read1_filenames.txt
+
+# list read 2 files and save output in a temporary file
+ls data/reads/*_2.fastq.gz > read2_filenames.txt
+
+# initiate a file with column names
+echo"fastq_1,fastq_2"> samplesheet.csv
+
+# paste the two temporary files together, using comma as a delimiter
+paste-d"," read1_filenames.txt read2_filenames.txt >> samplesheet.csv
+
+# remove the two temporary files
+rm read1_filenames.txt read2_filenames.txt
+
These commands resulted in creating a file called samplesheet.csv, which contains the following:
So, we programmatically created the last two columns of our file. We then opened this CSV file in Excel to add another column “sample” where we included our sample names and saved the file again as a CSV format. Here are the top few rows of the final file:
Now we are ready to run the nf-core/viralrecon pipeline (see Section 4.3 for details). We saved our command in a script (scripts/01-run_viralrecon.sh), which we created with the command line text editor nano. This ensures that our analysis is reproducible and traceable (we can go back to the script to see how the analysis was run).
+
First, we activate our software environment, to ensure Nextflow is available to us:
+
mamba activate nextflow
+
Then, we run our script with:
+
bash scripts/01-run_viralrecon.sh
+
Which will start executing the pipeline.
+
For reference, here is the command included in that script:
In the command above, you will notice several \ at the end of each line. This is indicating that we want to continue writing our command in the next line. Notice that the last line does not include \, because that is the end of the command. This is very useful when commands are very long, because it makes the code more readable.
+
+
+
After running the pipeline, we notice the following message:
+
-[nf-core/viralrecon] 2 samples skipped since they failed Bowtie2 1000 mapped read threshold:
+ 201: ZA09
+ 176: ZA12
+
This indicates that two samples - ZA09 and ZA12 - had very few reads aligned to the SARS-CoV-2 genome. Could the reason be that they had very few reads to start with? We can quickly investigate this hypothesis by counting the number of lines in the FASTQ files from these samples:
+
# FASTQ file for ZA09
+zcat data/reads/SRR17088930_1.fastq.gz |wc-l
+
698044
+
Since each sequence takes 4 lines in a FASTQ format, this indicates that this sample had 698044/4 = 124511 reads. That’s certainly more than 1000 reads, so the reason the sample failed must be something else. We will investigate this further in the next section.
+
+
+
+
9.4 Consensus Quality
+
+
+
+
+
+
+Note
+
+
+
+
See Section 5.2, if you need to revise how to assess the quality of consensus sequences.
+
+
+
+
9.4.1 General Metrics
+
We used the MultiqQC report to assess the initial quality of our samples. The quality report can be found in results/viralrecon/multiqc/multiqc_report.html.
+
We paid particular attention to:
+
+
Number of reads mapped to the reference genome.
+
Median depth of coverage.
+
Percentage of the genome with missing bases (‘N’).
+
Number of SNP + Indel variants.
+
+
+
+
+Example MultiQC output, highlighting the main columns with quality information that will be collected for the final report. Note that some columns have been ommited here for clarity.
+
+
+
We noted that:
+
+
2 samples - ZA09 and ZA12 - completely failed. This was already noted after we ran the pipeline, above. The reason seems to be because these samples have a very low percentage of non-human reads (less than 2%), indicating that the sequenced material was mostly human. Because of this, there were not enough reads to assemble a genome.
+
2 other samples - ZA10 and ZA23 - had > 90% missing bases, also indicating a very poor assembly. The reason was the same as above, both samples had low % of non-human reads.
+
1 samples - ZA14 - had ~39% missing bases. In this case the reason was not a low number of mapped reads. For example, sample ZA13 had a very similar median depth of coverage (121 vs 161 in ZA14) and even fewer mapped reads (24k vs 60k in ZA14). But ZA13 only had 2% missing bases. However, upon inspection of the amplicon heatmap, we detected that several amplicons were not properly amplified in ZA14 compared to ZA13. Therefore, the reason for high % missing bases in ZA14 was due to low amplification efficiency in this sample.
+
There was some systematic dropout for some amplicons, in particular nCoV-2019_64 had very low amplification in several of the samples. Of note was also nCoV-2019_73, and other neighbouring amplicons.
+
+
+
+
+Amplicon depth of sequencing (or coverage) across samples, from the MultiQC report. Darker regions indicate systematic amplicon dropout, probably due to issues during the PCR amplification.
+
+
+
Besides the MultiQC report, the pipeline also outputs a CSV file with collected summary metrics (equivalent to the first table on the report): results/viralrecon/multiqc/summary_variants_metrics_mqc.csv. We will use this file later to join this information with our metadata and lineage assignment using the R software (detailed in “Integration & Visualisation” section, below).
+
+
+
9.4.2 Variants
+
We also looked at the table of variants obtained from the pipeline. This is output in results/viralrecon/variants/ivar/variants_long_table.csv. This table can be very useful to keep track of particular mutations that may be increasing over time. Later, we will tidy this table to attach to our reported results (“Integration & Visualisation” section).
+
But for now, we will explore this table to address a few more quality-related questions. We opened this table in Excel to answer the following:
+
+
Where there samples with a high number of intermediate allele frequencies? This could indicate mixed samples due to cross-contamination.
+
+
Where there samples with frameshift mutations? These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.
+
+
By manual inspection of this table (and using the “filter” feature in Excel), we found 74 variants with alternative allele frequency less than 75%. These were spread across samples, all samples having less than 10 such low-frequency mutations, and most samples having less than 5. Sample ZA23 - which we had previously highlighted has having a high % missing bases (39%) - had 8 low-frequency mutations out of a total of 48 mutations in this sample (~16%), suggesting further quality issues in this sample.
+
+
+
+Variants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases.
+
+
+
+
+
+
9.5 Clean FASTA
+
The pipeline outputs the consensus sequences in results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa (one file for each sample). For downstream analysis, it is convenient to combine all these sequences into a single file, and also clean the sequence names (to remove some text – ” MN908947.3” –, which is added by the bcftools variant caller).
+
We created a new script to clean our consensus FASTA files, which we ran with bash scripts/02-clean_fasta.sh:
+
+
# combine and clean FASTA files
+cat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa |sed's| MN908947.3||'> report/consensus.fa
+
This command does two things:
+
+
Combine all our FASTA consensus sequences into a single file (using cat).
+
Clean the sequence names (using sed).
+
+
The output was saved as a new FASTA file: report/consensus.fa.
+
+
+
9.5.1 Missing Intervals
+
As a further quality check, we also generated a table of missing intervals (indicated by the N character in the FASTA sequences). We used the seqkit software to achieve this.
+
First, we activate our software environment:
+
mamba activate seqkit
+
Then, we ran the script bash scripts/03-missing_intervals.sh, which includes the following command:
This software outputs a tab-delimited table, which we saved as results/missing_intervals.tsv. The table looks like this (only the top few rows are shown):
We opened this file missing_intervals.tsv in Excel and quickly calculated the length of each interval. We noted that two samples - ZA10 and ZA23 - both have a continuous interval of over 18kb missing bases, which is not surprising as we had already identified these samples has having >90% missing data. We make a note of these samples as being possibly problematic in downstream analysis steps.
+
+
+
+
9.6 Downstream Analyses
+
Based on the clean consensus sequences, we then perform several downstream analysis.
+
+
9.6.1 Lineage Assignment
+
+
+
+
+
+
+Note
+
+
+
+
See Section 6.1, if you need to revise how lineage assignment works.
+
+
+
Although the Viralrecon pipeline runs Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). An up-to-date run of both of these tools can be done using each of their web applications:
However, for automation, reproducibility and traceability purposes, we used the command line versions of these tools, and included their analysis in two scripts.
For Nextclade, we first activate the software environment:
+
mamba activate nextclade
+
And then we ran the script bash scripts/04-nextclade.sh, which contains the following commands:
+
# get nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa
+
The first command downloads the latest version of the Nextclade background data using nextclade dataset. We use that data as input to the second command (nextclade run) to make sure it runs with the most up-to-date lineages.
+
+
+
For Pangolin, we first activate the software environment:
+
mamba activate pangolin
+
And then we ran the script bash/04-pangolin.sh, which contains the following commands:
+
# update pangolin data
+pangolin--update-data
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa
+
Similarly to before, we first ran pangolin --update-data to ensure we were using the latest lineages available. We can check the version of the data used with pangolin --all-versions (at the time we ran this we had pangolin-data: 1.23.1).
+
+
+
+
Both of these tools output CSV files, which can be open in Excel for further examination.
+Opening the pangolin results (results/pangolin/pango_report.csv), we noticed that the two problematic samples – ZA10 and ZA23 – failed the QC due to high fraction of missing data. The other samples all seemed to have been assigned to “Omicron” variant with a high support.
+
Opening the nextclade results (results/nextclade/nextclade.tsv), we noticed that the two problematic samples were classified as “recombinant”! We know from our quality control that we should not trust this assessment, and that most likely these are bad quality samples, not true recombinant lineages. Nextclade is more relaxed in assigning samples to lineages, so we should always check the QC status as well. We will notice that both of these samples were assigned QC status “bad”, due to their high percentage of missing data (nextclade uses a stringent threshold of 3000 sites, or ~10%, missing data). Two other samples had “bad” QC status. Sample ZA14 due to high % of missing data, and sample ZA18 from a mixture of a high number of private mutations and the presence of a frameshift mutation in ORF1b.
+
Like before, we will do further analysis (and visualisation) of these data using the software R, in the section “Integration & Visualisation”, detailed below.
+
+
+
9.6.2 Phylogeny
+
+
+
+
+
+
+Note
+
+
+
+
See Section 7.1, if you need to revise how to build phylogenetic trees.
+
+
+
Although a tool such as Nextclade can place our samples in a global phylogeny context, sometimes it may be convient to build our own phylogenies. This requires three steps:
+
+
Producing a multiple sequence alignment from all consensus sequences.
+
Tree inference.
+
Tree visualisation and annotation.
+
+
Before our analysis, we first activated our software environment:
+
mamba activate phylo
+
We performed the first two steps with the following script, which we ran with bash scripts/05-phylogeny.sh:
The output of iqtree includes a tree file, which can be visualised using FigTree (or online using Microreact). The figure below shows our tree, which shows all our samples fall mostly clustering together. This makes sense, as all our samples were classified as “Omicron (BA.1-related)”.
+
It is worth noting that the samples ZA10, ZA14 and ZA23 are not included in this phylogeny, as they contained >20% missing data (we used that threshold with MAFFT alignment, option --maxambiguous 0.2). Also, note that sample ZA18, which Nextclade had identified as “bad” QC (due to excess private mutations) also appears slightly separated from the other samples in the tree (the sample in red on the tree). The sample still clusters well with the rest, suggesting we can probably trust that it is indeed an Omicron variant, however the excess of private mutations (which may be due to sequencing errors) is making it stand apart from the others in the phylogeny.
+
+
+
+Phylogenetic tree for our samples. All samples were classified as lineage BA.1 (and some of its sub-lineages), corresponding to the Omicron VOC. The tips of the tree are coloured according to Nextclade’s QC status: green = “good”; blue = “mediocre”; red = “bad”.
+
+
+
+
+
9.6.3 Clustering
+
We identified groups of similar sequences in our data using the software civet (Cluster Investigation and Virus Epidemiology Tool). This software compares our samples with a background dataset of our choice, which givus us more context for our analysis. In this case we are using the example background data that comes with civet. However, in a real-world analysis, it would have been ideal to choose local samples as background data. For example, we could download samples from South Africa from around the time period of our sample collection, from GISAID following the instructions on the civet documentation (you need an account on GISAID to obtain these data).
+
For this example, we already prepared civet background dataset saved in resources/civet_background_data.
+
Before our analysis, we first activate our software environment:
+
mamba activate civet
+
Then, we ran the script bash scripts/06-civet.sh, which contains the following code:
The result of this analysis includes an interactive HTML report (in results/civet/civet.html). We can see that our samples were grouped into a single catchment. This makes sense from our previous lineage/variant analysis: all our samples were classfied as Omicron VOC. We can see that our samples seem to be more diverged from the Australian BA.1 sample present in the background data, with new SNPs in our samples creating a longer branch in the tree.
+
+
+
+Results from civet report for catchment 1, showing the phylogeny in the context of a BA.1 Australian sample from the background data.
+
+
+
Civet also outputs a CSV file (results/civet/master_metadata.csv), which includes the catchment that each sample was assigned to. We will use this CSV file later to integrate this information with other parts of our analysis, in R, detailed in the “Integration & Visualisation” section.
+
+
+
+
9.7 Integration & Visualisation
+
At this point in our analysis, we have several tables with different pieces of information:
+
+
sample_info.csv → the original table with metadata for our samples.
+
results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.
+
results/nextclade/nextclade.tsv → the results from Nextclade.
+
results/pangolin/pango_report.csv → the results from Pangolin.
+
results/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.
+
+
To consolidate our analysis, we tidied and integrated the information from across these different files, into a single table using the software R. The script used to do this is in scripts/07-data_integration.R. Because this is an R script, we opened it in RStudio to execute the code.
+
The output of our script is a new tab-delimited table, which we saved in report/consensus_metrics.tsv, and contains the following columns:
+
+
sample → sample ID.
+
collection_date → date of collection day.
+
collection_week → date of collection week (useful for summarising/visualising counts per-week).
+
country → country of origin.
+
latitude/longitude → latitude and longitude of collection.
+
n_mapped_reads → number of mapped reads.
+
median_depth → median depth.
+
pct_missing → percentage of missing data.
+
pct_coverage → percentage of coverage.
+
n_variants → number of SNP + indel variants detected.
+
nextclade → nextclade clade.
+
qc_status → QC status as determined by Nextclade (“bad”, “mediocre”, “good”).
+
lineage → Pangolin lineage.
+
who_variant → variant of concern designation.
+
catchment → catchment group from Civet.
+
+
This table, which aggregates information from many of the tools we used, was then used to produce different visualisations of our analysis. These visualisations were also done using the R software (scripts/08-visualisation.R), and integrated into a report, shown below.
+
+
+
+
9.8 Bonus: Full Workflow
+
Although we have ran each of the steps of our analysis individually (each in their own script), now that we have everything working, we could integrate all these steps into a single “master” script:
+
#!/bin/bash
+
+# make mamba activate command available
+eval"$(conda shell.bash hook)"
+source$(mamba info --base)/etc/profile.d/mamba.sh
+
+# make report directory
+mkdir-p report
+
+mamba activate nextflow
+# run viralrecon
+nextflow run nf-core/viralrecon \
+-r 2.6.0 -profile singularity \
+--max_memory'15.GB'--max_cpus 8 \
+--platform illumina \
+--input samplesheet.csv \
+--outdir results/viralrecon \
+--protocol amplicon \
+--genome'MN908947.3'\
+--primer_set artic \
+--primer_set_version 3 \
+--skip_assembly--skip_asciigenome\
+--skip_pangolin--skip_nextclade
+
+# combine and clean FASTA files
+cat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa |sed's| MN908947.3||'> report/consensus.fa
+
+mamba activate seqkit
+# create missing bases TSV file
+seqkit locate -i-P-G-M-r-p"N+" report/consensus.fa > results/missing_intervals.tsv
+
+mamba activate nextclade
+# get nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa
+
+mamba activate pangolin
+# update pangolin data
+pangolin--update-data
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa
+
+mamba activate phylo
+# alignment
+mkdir-p results/mafft
+mafft--6merpair--maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa
+
+# tree inference
+mkdir-p results/iqtree
+iqtree2-s results/mafft/alignment.fa --prefix results/iqtree/consensus
+
+# data integration and cleaning
+Rscript scripts/07-data_integration.R
+
Notice that we included the R script that does the data cleaning here, using the Rscript program that allows to execute an R script from the command-line.
+
Having this “master” script, we could run all these steps from start-to-finish with a single command, which can be very useful if you want to fully automate your analysis across multiple runs.
This section is an extended self-paced practice applying the concepts covered in previous sections.
+
By the end of this practice, you should be able to:
+
+
Prepare all the files necessary to run the consensus pipeline.
+
Run the viralrecon pipeline to generate FASTA consensus from raw FASTQ files.
+
Assess and collect several quality metrics for the consensus sequences.
+
Clean output files, in preparation for other downstream analysis.
+
Assign sequences to lineages.
+
Contextualise your sequences in other background data and cluster them based on phylogenetic analysis.
+
Integrate the metadata and analysis results to generate timeseries visualisations of your data.
+
+
+
+
+
External Quality Assessment (EQA) is a procedure that allows laboratories to assess the quality of the data they produce, including its analysis. For genomic analysis of SARS-CoV-2, a standard panel of samples has been developed by the GenQA consortium, which is part of UK NEQAS, a non-profit that designs EQA protocols for a range of applications. GenQA’s panel of samples includes lab-cultured SARS-CoV-2 samples of known origin, enabling laboratories to assess whether their sequencing and bioinformatic pipelines correctly identify expected mutations and lineage assignments for each of the samples in the panel.
+
+
+
+
+
+
+More about EQA
+
+
+
+
+
+
EQA programs such as this one help to provide assurance of the diagnostic testing results obtained by a lab. One of the key things to consider is that, for reliable assessment, samples should be processed in the same way as patient samples. Since these samples are for quality assessment, it may be tempting to assign the samples to more experienced staff, include extra checks, increase sequencing, etc. However, doing that would give a false impression of the performance of your own lab.
+
Evaluating the results of EQA sample processing is critical for labs to understand where their procedures can be improved. This may include staff training, purchase of new equipment, or updating the reagents and kits used for sample processing.
+
EQA panels are often updated, to reflect any new and emerging pathogens, allowing the labs to assess the suitability of the protocols used to detect them. It also helps raise awareness of any atypical variants of pathogens that may exist in the environment.
+
+
+
+
In this case study, we are going to analyse samples from the GenQA panel, to helps us assess the quality of our bioinformatic analysis and extract key pieces of information to be reported. Two panels are available, which include the following variants:
From the sequencing data analysis, we will address the following:
+
+
What was the quality of the consensus sequence obtained?
+
What lineage/clade was each of our samples assigned to? Did we identify the correct (expected) lineage?
+
Which of the expected mutations were we able to detect? Where there any “false positives” or “false negatives”?
+
+
We should also produce several essential output files, which would usually be necessary to upload our data to public repositories:
+
+
Consensus sequences (FASTA)
+
Metadata and consensus sequence quality metrics (TSV)
+
Variants (TSV)
+
+
+
10.1 Pipeline Overview
+
We will start our analysis with FASTQ files produced by our sequencing platforms (Illumina and Nanopore are considered here). These FASTQ files will be used with the nf-core/viralreconNextflow pipeline, allowing us to automate the generation of consensus sequences and produce several quality control metrics essential for our downstream analysis and reporting.
+
Critical files output by the pipeline will need to be further processed, including combining and cleaning our consensus FASTA files. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions.
+
+
+
+
10.2 Preparing Files
+
Before we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. There is no standard way to do this, but here are some suggested directories:
+
+
data → contains the sequencing data for the particular run or project you are working on. Data may sometimes be stored in a separate server, in which case you may not need to create this directory. Generally, you should leave the original data unmodified, in case something goes wrong during your analysis, you can always re-run it from the start.
+
results → to save results of the analysis.
+
scripts → to save scripts used to run the analysis. You should always try to save all the commands you run in a script, this way you will have a record of what was done to produce the results, and it makes your life easier if you want to run the analysis again on a new set of data.
+
report → to save the final files and documents that we report to our colleagues or upload to public repositories.
+
resources → files that you download from the internet could be saved here. For example, the reference genome sequence, background data used for phylogenetics, etc. Sometimes you may share these resources across multiple projects, in which case you could have this folder somewhere else that you can access across multiple projects.
+
+
On our computers, we have a directory in ~/Documents/eqa_workshop, where we will do all our analysis. We already include the following:
+
+
data → with the results of the EQA sample sequencing.
+
resources → where we include the SARS-CoV-2 reference genome, and some background datasets that will be used with some of the tools we will cover.
+
scripts → where we include some scripts that we will use towards the end of the workshop. You should also create several scripts during the workshop, which you will save here.
+
sample_info.csv → a table with some metadata for our samples.
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Your first task is to create two new directories in the project folder called report and results. You can do this either using the file explorer or from the command line (using the mkdir command).
+
+
+
+
+
+
+
10.2.1 Data
+
Regardless of which platform you used to sequence your samples, the analysis starts with FASTQ files (if you need a reminder of what a FASTQ file is, look at the Introduction to NGS section). However, the organisation of these files is slightly different depending on the platform, and is detailed below.
Typically, Nanopore data is converted to FASTQ format using the program Guppy. This software outputs the files to a directory called fastq_pass. Within this directory, it creates further sub-directories for each sample barcode, which are named barcodeXX (XX is the barcode number). Finally, within each barcode directory there is one (or sometimes more) FASTQ files corresponding to that sample.
+
You can look at the files you have available from the command line using:
+
ls data/fastq_pass
+
+
+
The Illumina files come as compressed FASTQ files (.fq.gz format) and there’s two files per sample, corresponding to read 1 and read 2. This is indicated by the file name suffix:
+
+
*_1.fq.gz for read 1
+
*_2.fq.gz for read 2
+
+
You can look at the files you have available from the command line using:
+
ls data/
+
+
+
+
+
+
10.2.2 Metadata
+
A critical step in any analysis is to make sure that our samples have all the relevant metadata associated with them. This is important to make sense of our results and produce informative reports at the end. There are many types of information that can be collected from each sample (revise the Genomic Surveillance > Metadata section of the materials to learn more about this). For effective genomic surveillance, we need at the very minimum three pieces of information:
+
+
When: date when the sample was collected (not when it was sequenced!).
+
Where: the location where the sample was collected (not where it was sequenced!).
+
How: how the sample was sequenced (sequencing platform and protocol used).
+
+
Of course, this is the minimum metadata we need for a useful analysis. However, the more information you collect about each sample, the more questions you can ask from your data – so always remember to record as much information as possible for each sample.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Note: If you are using our pre-sequenced data, you can skip this exercise.
+
We already provide some basic metadata for these samples in the file sample_info.csv:
+
+
sample → the sample ID.
+
collection_date → the date of collection for the sample in the format YYYY-MM-DD.
+
country → the country of origin for this sample.
+
expected_lineage and expected_voc → because we are using the EQA panel, we know what lineage and variant-of-concern (VOC) each sample belongs to. We will use this later to assess the quality of our analysis.
+
+
There is however some information missing from this table, which you may have available at this point. Open this file in Excel and create the following columns:
+
+
ct → Ct value from qPCR viral load quantification.
+
sequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).
+
sequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).
+
amplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)
+
Specific columns for Oxford Nanopore data, which are essential for the bioinformatic analysis:
+
+
ont_nanopore → the version of the pores used (e.g. 9.4.1 or 10.4.1).
+
ont_guppy_version → the version of the Guppy software used for basecalling.
+
ont_guppy_mode → the basecalling mode used with Guppy (usually “fast”, “high”, “sup” or “hac”).
+
+
+
This will ensure that in the future people have sufficient information to re-run the analysis on your data.
+
+
+
+
+
+
+
+
+
+
+
+Dates in Spreadsheet Programs
+
+
+
+
Note that programs such as Excel often convert date columns to their own format, and this can cause problems when analysing data later on. For example, GISAID wants dates in the format YYYY-MM-DD, but by default Excel displays dates as DD/MM/YYYY.
+You can change how Excel displays dates by highlighting the date column, right-clicking and selecting Format cells, then select “Date” and pick the format that matches YYYY-MM-DD. However, every time you open the CSV file, Excel annoyingly converts it back to its default format!
+
To make sure no date information is lost due to Excel’s behaviour, it’s a good idea to store information about year, month and day in separate columns (stored just as regular numbers).
+
+
+
+
+
+
10.3 Consensus Assembly
+
At this point we are ready to start our analysis with the first step: generating a consensus genome for our samples. We will use a standardised pipeline called viralrecon, which automates most of this process for us, helping us be more efficient and reproducible in our analysis.
+
+
+
+
+
+
+Note
+
+
+
+
See Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.
+
+
+
+
10.3.1 Samplesheet
+
The first step in this process is to prepare a CSV file with information about our sequencing files, which will be used as the input to the viralrecon pipeline.
+The pipeline’s documentation gives details about the format of this samplesheet, depending on whether you are working with Illumina or Nanopore data.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Note: If you are using our pre-sequenced data, you can skip this exercise.
+
Using Excel, produce the input samplesheet for nf-core/viralrecon, making sure that you save it as a CSV file (File → Save As… and choose “CSV” as the file format).
+
Note: make sure that the sample names you use in this samplesheet match those in the metadata sample_info.csv file (pay attention to things like spaces, uppercase/lowercase, etc. – make sure the names used match exactly).
+
If you are working with Illumina data, you should check the tip below.
+
+
+Illumina samplesheet: saving time with the command line!
+
+
You can save some time (and a lot of typing!) in making the Illumina samplesheet using the command line to get a list of file paths. For example, if your files are saved in a folder called data, you could do:
+
# list read 1 files and save output in a temporary file
+ls data/*_1.fq.gz > read1_filenames.txt
+
+# list read 2 files and save output in a temporary file
+ls data/*_2.fq.gz > read2_filenames.txt
+
+# initiate a file with column names
+echo"fastq_1,fastq_2"> samplesheet.csv
+
+# paste the two temporary files together, using comma as a delimiter
+paste-d"," read1_filenames.txt read2_filenames.txt >> samplesheet.csv
+
+# remove the two temporary files
+rm read1_filenames.txt read2_filenames.txt
+
Now, you can open this file in Excel and continue editing it to add a new column of sample names.
+
+
+
+
+
+
+
+
+
10.3.2 Running Viralrecon
+
The next step in our analysis is to run the nf-core/viralrecon pipeline. The way the command is structured depends on which kind of data we are working with. There are many options that can be used to customise the pipeline, but typical commands are shown below for each platform.
You need to check which model the medaka software should use to process the data. You need three pieces of information to determine this:
+
+
The version of the nanopores used (usually 9.4.1 or 10.4.1).
+
The sequencing device model (MinION, GridION or PromethION).
+
The mode used in the Guppy software for basecalling (“fast”, “high”, “sup” or “hac”).
+
The version of the Guppy software.
+
+
Once you have these pieces of information, you can see how the model is specified based on the model files available on the medaka GitHub repository. For example, if you used a flowcell with chemistry 9.4.1, sequenced on a MinION using the “fast” algorithm on Guppy version 5.0.7, the model used should be r941_min_fast_g507.
+Note that in some cases there is no model for recent versions of Guppy, in which case you use the version for the latest version available. In our example, if our version of Guppy was 6.1.5 we would use the same model above, since that’s the most recent one available.
Your next task is to run the pipeline on your data. However, rather than run the command directly from the command line, let’s save it in a shell script – for reproducibility and as a form of documenting our analysis.
+
+
First, open the README.txt file found in your folder, which has some details about your samples, including the amplicon primer scheme used.
+
Using a text editor, create a new shell script and save it in scripts/01-run_viralrecon.sh. You can either use the command-line text editor nano or use Gedit, which comes installed with Ubuntu.
+
Copy the command shown above (either Illumina or Nanopore, depending on your data) to your new script.
+
Adjust the code, to use the correct input samplesheet file, primer scheme and, in the case of ONT, the medaka model. For information about primer scheme options see the “Amplicon Primer Schemes” appendix.
+
Activate the software environment to use Nextflow: mamba activate nextflow.
+
Save the script and run it from the command line using bash scripts/01-run_viralrecon.sh.
+
+
If you need a reminder of how to work with shell scripts, revise the Shell Scripts section of the accompanying Unix materials.
+
+
+
+
+
+
+
+
+
+
+
+Maximum Memory and CPUs
+
+
+
+
In our Nextflow command above we have set --max_memory '15.GB' --max_cpus 8 to limit the resources used in the analysis. This is suitable for the computers we are using in this workshop. However, make sure to set these options to the maximum resources available on the computer where you process your data.
+
+
+
+
+
+
10.4 Consensus Quality
+
Once your workflow is complete, it’s time to assess the quality of the assembly.
+
+
+
+
+
+
+Note
+
+
+
+
See Section 5.2, if you need to revise how to assess the quality of consensus sequences.
+
+
+
+
10.4.1 Coverage
+
At this stage we want to identify issues such as:
+
+
Any samples which have critically low coverage. There is no defined threshold, but samples with less than 85% coverage should be considered carefully.
+
Any problematic regions that systematically did not get amplified (amplicon dropout).
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
To assess the quality of our assemblies, we can use the MultiQC report generated by the pipeline, which compiles several pieces of information about our samples. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.
+
Open the quality report and try to answer the following questions:
+
+
Were there any samples with more than 15% missing bases (’N’s)?
+
Were there any samples with a median depth of coverage <20x?
+
Were there any problematic amplicons with low depth of coverage across multiple samples?
+
+
Make a note of any samples that you think are problematic. You can discuss with your colleagues and compare your results/conclusions to see if you reach similar conclusions.
+
+
+
+
+
+
+
+
10.4.2 Variants
+
The viralrecon pipeline outputs a table with information about SNP/Indel variants as a CSV file named variants_long_table.csv. It is important to inspect the results of this file, to identify any mutations with severe effects on annotated proteins, or identify samples with an abnormal high number of “mixed” bases.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Open the variants_long_table.csv file and answer the following questions. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.
+
+
How many variants have allele frequency < 75%?
+
Does any of the samples have a particularly high number of these low-frequency variants, compared to other samples? (This could indicate cross-contamination of samples)
+
Investigate if there are any samples with frameshift mutations. These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.
Make a note of any samples that you think may be problematic, either because they have a high number of low-frequency variants or because they have frameshift mutations.
+
(Optional)
+If you identify samples with frameshift mutations, open the BAM file of the sample using the software IGV. Go to the position where the mutation is located, and try to see if there is evidence that this mutation is an error (for example, if it’s near an homopolymer).
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
The samples we are using in this analysis come from a standard EQA panel. As such, we already know what lineages we expect to find in these samples.
+
+
Open IGV and load one (or more) of the alignment BAM files into it. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.
+
Open your metadata table (sample_info.csv) and check which lineages your loaded samples belong to.
+
Do a web search to find what mutations characterise those lineages.
+
On IGV, go to the location of some of the expected mutations, and see if you can see it in the respective samples.
+
+
+
+
+
+
+
+
+
10.4.3 Clean FASTA
+
The viralrecon pipeline outputs each of our consensus sequences as individual FASTA files for each sample (look at the FASTA Files section if you need a reminder of what these files are). However, by default, the sample names in this file have extra information added to them, which makes some downstream analysis less friendly (because the names will be too long and complicated).
+
To clean up these files, we will do two things:
+
+
Combine all the individual FASTA files together.
+
Remove the extra text from the sequence names.
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
We will use the programs cat (concatenate) and sed (text replacement) to clean our FASTA files. Consider the commands given below:
The sed command shown is used to remove the extra pieces of text added by the pipeline to each sample name. This is a slightly more advanced program to use, so we already give you the code. If you want to learn more about it, check the “Text Replacement” section of the accompanying Unix materials.
+
Copy the command above to a new shell script called scripts/02-clean_fasta.sh. Fix the code by replacing <INPUT> with the path to all the FASTA files generated by the pipeline (remember you can use the * wildcard).
+If you need a reminder of where to find the FASTA consensus files from viralrecon, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.
+
Once you fixed the script, run it with bash and check that the output file is generated.
+
Bonus:
+Check that you have all expected sequences in your FASTA file using grep to find the lines of the file with > (which is used to indicate sample names).
+
+
+
+
+
+
+
+
10.4.4 Missing Intervals
+
When we generate our consensus assembly, there are regions for which we did not have enough information (e.g. due to amplicon dropout) or where there were mixed bases. These regions are missing data, and are denoted as ‘N’ in our sequences. Although we already determined the percentage of each sequence that is missing from the MultiQC report, we don’t have the intervals of missing data, which can be a useful quality metric to have. In particular, we may want to know what is the largest continuous stretch of missing data in each sample.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
We will use the software tool seqkit to find intervals where the ‘N’ character occurs. This tool has several functions available, one of which is called locate.
+
Here is the command that would be used to locate intervals containing one or more ‘N’ characters:
Create a new shell script called scripts/03-missing_intervals.sh.
+
Copy the command shown above to the script and modify it to redirect the output (using >) to a file called results/missing_intervals.tsv.
+
Activate the software environment: mamba activate seqkit.
+
Run the script you created using bash.
+
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Open the file you created in the previous step (results/missing_intervals.tsv) in a spreadsheet program. Create a new column with the length of each interval (end - start + 1).
+
Note if any missing intervals are larger than 1Kb, and whether they overlap with the Spike gene. (Note: you can use the Sars-CoV-2 genome browser to help you see the location of the annotated genes.)
+
+
+
+
+
+
+
+
+
10.5 Downstream Analyses
+
Now that we have cleaned our FASTA file, we can use it for several downstream analysis. We will focus on these:
+
+
Lineage assignment: identify if our samples come from known lineages from the Pangolin consortium.
+
Phylogeny: produce an annotated phylogenetic tree of our samples.
+
Clustering: assess how many clusters of sequences we have, based on a phylogenetic analysis.
+
Integration & Visualisation: cross-reference different results tables and produce visualisations of how variants changed over time.
+
+
+
10.5.1 Lineage Assignment
+
+
+
+
+
+
+Note
+
+
+
+
See Section 6.1, if you need to revise how lineage assignment works.
+
+
+
Although the Viralrecon pipeline can run Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). Although it is possible to configure viralrecon to use more recent versions of these tools, it requires more advanced use of configuration files with the pipeline.
+
Alternatively, we can run our consensus sequences through the latest versions of Nextclade and Pangolin. There are two ways to do this: using their respective web applications, or their command-line versions. For this task, we recommend that you use the command line tools (this will ensure our downstream analysis works well), but we also provide the option to use the web apps for your reference.
To run the command-line version of these tools, there are two steps:
+
+
Update the datasets of each software (to ensure they are using the latest lineage/clade nomenclature available).
+
Run the actual analysis on your samples.
+
+
We will use the exercises below to see what the commands to achieve this are.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
We will start doing the Nextclade analysis.
+
+
Create a new script file called scripts/04-nextclade.sh and copy this code into it:
+
# update nextclade data
+nextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data
+
+# run nextclade
+nextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade/ <INPUT>
+
Fix the code, replacing <INPUT> with the path to your consensus sequence file. Save the file.
+
Activate the software environment: mamba activate nextclade.
+
Run your script using bash.
+
Once the analysis completes, open the file results/nextclade/nextclade.tsv in Excel and see what problems your samples may have (in particular those classified as “bad” quality).
+
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
For the Pangolin analysis:
+
+
Create a new script file called scripts/04-pangolin.sh and copy this code into it:
+
# update pangolin data
+<DATA_UPDATE_COMMAND>
+
+# run pangolin
+pangolin--outdir results/pangolin/ --outfile pango_report.csv <INPUT>
+
Fix the code:
+
+
Replace <INPUT> with the path to your consensus sequence file.
+
Replace <DATA_UPDATE_COMMAND> with the pangolin command used to update its data. Check the documentation of the tool with pangolin --help to see if you can find what this option is called. Alternatively, look at the documentation online.
+
+
Save the file.
+
Activate the software environment: mamba activate pangolin.
+
Run your script using bash.
+
Once the analysis completes, open the file results/pangolin/pango_report.csv in Excel and see if there were any samples for which the analysis failed. If there were any failed samples, check if they match the report from Nextclade.
+
+
+
+
+
+
+
+
+
These exercises are optional - during the workshop please use the command line version of the tools.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Running Nextclade
+
Go to clades.nextstrain.org and run Nextclade on the clean FASTA file you created earlier (report/consensus.fa).
+If you need a reminder about this tool, see the “Lineages and variants” section of the materials.
+
Once the analysis completes, pay particular attention to the quality control column, to see what problems your samples may have (in particular those classified as “bad” quality).
+
Then:
+
+
Create a new folder in your project directory: results/nextclade.
+
Use the “download” button (top-right) and download the file nextclade.tsv (tab-delimited file), which contains the results of the analysis. Important: please download the TSV file (not the CSV file, as it uses ; to separate columns, and will not work later with the “Data Integration” section).
+
Save this file in the results/nextclade folder you created.
+
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Running Pangolin
+
Go to pangolin.cog-uk.io and run Pangolin on the clean FASTA file you created earlier (report/consensus.fa).
+If you need a reminder about this tool, see the “Lineages and variants” section of the materials.
+
Once the analysis completes, pay particular attention to any samples that failed. If there were any failed samples, check if they match the report from Nextclade.
+
Then:
+
+
Create a new folder in your project directory: results/pangolin.
+
Use the “download” button (the “arrow down” symbol on the results table) and download the file results.csv. Rename this file to report.csv (important: make sure to rename the file like this, otherwise it will not work later with the “Data Integration” section).
+
Save this file in the results/pangolin folder you created.
+
+
+
+
+
+
+
+
+
+
+
+
10.5.2 Phylogeny
+
+
+
+
+
+
+Note
+
+
+
+
See Section 7.1, if you need to revise how to build phylogenetic trees.
+
+
+
Although tools such as Nextclade and civet can place our samples in a phylogeny, sometimes it may be convenient to build our own phylogenies. This requires three steps:
+
+
Producing a multiple sequence alignment from all consensus sequences.
+
Tree inference.
+
Tree visualisation.
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
+
Start by creating two directories to store the output of our analysis: results/mafft and results/iqtree.
+
Activate the software environment: mamba activate phylo (this environment includes both mafft and iqtree).
+
To make our analysis more interesting, we will combine our sequences with sequences from previous workshops. Use the cat program to concatenate your sequences (report/consensus.fa) with the sequences from our collaborators (resources/eqa_collaborators/eqa_consensus.fa). Using >, save the output in a new file results/mafft/unaligned_consensus.fa.
+
Perform a multiple sequence alignment of the combined consensus sequences using the program mafft (see Section 7.2 for how to use this program). Save the output in a file called results/mafft/aligned_consensus.fa.
+
Infer a phylogenetic tree using the iqtree2 program (see Section 7.3).
+
Once you have both of these commands working, make sure to save them in a new shell script (as a record of your analysis). Save the script as scripts/05-phylogeny.sh.
+
Visualise the tree using FigTree (see Section 7.4).
+
+
What substitution model was chosen as the best for your data by IQ-Tree?
+
Do your samples group with samples from previous workshops in the way that is expected, or are there any outliers?
+
Are there any obvious differences between sequencing technologies?
+
+
+
+
+
+
+
+
10.5.3 Clustering (Optional Section)
+
+
The software civet (Cluster Investigation and Virus Epidemiology Tool) can be used to produce a report on the phylogenetic relationship between viral samples, using a background dataset to contextualise them (e.g. a global or regional sample of sequences) as well as metadata information to enable the identification of emerging clusters of samples.
+
Civet works in several stages, in summary:
+
+
Each input sequence is assigned to a “catchment” group. These are groups of samples that occur at a given genetic distance (number of SNP differences) from each other.
+
Within each catchment group, a phylogeny is built (using the iqtree2 software with the HKY substitution model).
+
The phylogeny is annotated with user-input metadata (e.g. location, collection date, etc.). The results are displayed in an interactive HTML report.
+
+
One of the critical pieces of information needed by civet is the background data, to which the input samples will be compared with. These background data could be anything the user wants, but typically it’s a good idea to use samples from the local geographic area from where the samples were collected. For example, a national centre may choose to have their background data composed of all the samples that have been sequenced in the country so far. Or perhaps all the samples in its country and other neighbouring countries.
+
One way to obtain background data is to download it from GISAID. An example of how to do this is detailed in the civet documentation. We have already downloaded the data from GISAID, so we can go straight to running our civet analysis.
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
+
Create a new script in scripts/06-civet.sh, with the following command, adjusted to fit your files:
Activate the software environment: mamba activate civet.
+
Once your script is ready, run it with bash.
+
After the analysis completes, open the HTML output file in results/civet and examine into how many catchments your samples were grouped into.
+
+
+
+
+
+
+
+
+
+
10.6 Integration & Visualisation
+
At this point in our analysis, we have several tables with different pieces of information:
+
+
sample_info.csv → the original table with metadata for our samples.
+
results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.
+
results/nextclade/nextclade.tsv → the results from Nextclade.
+
results/pangolin/pango_report.csv → the results from Pangolin.
+
(optional) results/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.
+
+
Each of these tables stores different pieces of information, and it would be great if we could integrate them together, to facilitate their interpration and generate some visualisations.
+
We will demonstrate how this analysis can be done using the R software, which is a popular programming language used for data analysis and visualisation. Check the Quick R Intro appendix for the basics of how to work with R and RStudio.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Data Integration
+
From RStudio, open the script in scripts/07-data_integration.R. Run the code in the script, going line by line (remember in RStudio you can run code from the script panel using Ctrl + Enter). As you run the code check the tables that are created (in your “Environment” panel on the top-right) and see if they were correctly imported.
+
Once you reach the end of the script, you should have two tab-delimited files named report/consensus_metrics.tsv and report/variants.tsv. Open both files in Excel to check their content and confirm they contain information from across the multiple tables.
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Data Visualisation
+
After integrating the data, it’s time to produce some visualisations. From RStudio, open the script in scripts/08-visualisation.R. Run the code in the script, going line by line.
+
As you run the code, several plots will be created. You can export these plots from within RStudio using the “Export” button on the plotting panel – these will be useful when you write your report later.
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Annotating Phylogenetic Tree
+
Using FigTree, import two annotation files (File > Import Annotations…):
+
+
report/consensus_metrics.tsv, which was created in the Data Integration exercise.
+
resources/eqa_collaborators/metadata.tsv, which has lineage assignment for EQA samples sequenced by other labs.
+
+
After importing both files, annotate your phylogenetic tree to display the lineages assigned to each sample as the tip labels. See Section 7.4, if you need a reminder of how to annotate trees using FigTree.
+
+
+
+
+
+
+
+
10.7 EQA Panels
+
The analysis we have done so far is applicable to any new sequences that you want to process. However, we have also been using a pre-defined panel of samples to be used as part of an External Quality Assessment (EQA) process. This allows us to assess whether our bioinformatic analysis identified all the expected mutations in these samples, as well as assigned them to the correct lineages.
+
To do this, requires to compare the mutations we detected in our EQA samples to the expected mutations provided to us. From this, we will be able to calculate True Positives (TP), False Positives (FP) and False Negatives (FN), and calculate two performance metrics:
+
+
Precision = TP / (TP + FP) → what fraction of the mutations we detected are true?
+
Sensitivity = TP / (TP + FN) → what fraction of all true mutations did we detect?
+
+
There can be cases where one of these metrics is high and the other one lower. For example, if you have a high-coverage genome (say >95%) but lots of sequencing errors, you may have a high sensitivity (you manage to detect all true mutations), but low precision (you also detect lots of false positives, due to sequencing errors). Conversely, if you have a low-coverage genome (say <50%) but very high-quality sequencing, you may have low sensitivity (you missed lots of true mutations, because of missing data), but high precision (those mutations that you did manage to identify were all true, you didn’t have many false positives).
+
+
If you are submiting your samples to GenQA’s platform, they will also provide with a general accuracy score called F-score. This is calculated as the harmonic mean of precision and sensitivity:
A high F-score is indicative of both high precision and sensitivity, whereas a lower score indicates that at least one of those metrics are low.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
From RStudio, open the script in scripts/09-eqa_validation.R. Run the code in the script, going line by line.
+
This script will save a new tab-delimited file in report/eqa_performance_metrics.tsv. Open this file in Excel and check what the precision and sensitivity of your analysis was. Are you happy with the results?
+
+
+
+
+
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
(Optional)
+
While you run the R script, you will have an object called all_mutations in your environment (check the top-right panel). From RStudio, click on the name of this object to open it in the table viewer.
+
Look at mutations that were not detected in your samples, but were present in the expected mutations (i.e. false negatives). Open the BAM file for one of those samples in IGV and navigate to the position where that mutation should occur. Investigate what the reason may have been for missing that mutation.
Finally, we have come to the end of our analysis. So, all that is left is to report our results in the most informative way to our colleagues and decision-makers. You may already have established reporting templates in your institution, and if that’s the case, you should use those. Alternatively, we will look at a suggested template, based on the reports done by the UK Health Security Agency (UKHSA).
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
Open our shared report template document and download it as a Word document (File → Download → Microsoft Word (.docx)). Save the file in report/YYYY-MM-DD_analysis_report.docx (replace YYYY-MM-DD by today’s date).
+
Open the file and complete it with the information you collected throughout your analysis. You should be able to get all this information from the files in your report/ directory.
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
\ No newline at end of file
diff --git a/materials/03-case_studies/images/analysis_overview.png b/materials/03-case_studies/images/analysis_overview.png
new file mode 100644
index 0000000..81f4df9
Binary files /dev/null and b/materials/03-case_studies/images/analysis_overview.png differ
diff --git a/materials/03-case_studies/images/civet_catchment_example.png b/materials/03-case_studies/images/civet_catchment_example.png
new file mode 100644
index 0000000..efb3056
Binary files /dev/null and b/materials/03-case_studies/images/civet_catchment_example.png differ
diff --git a/materials/03-case_studies/images/precision_sensitivity.svg b/materials/03-case_studies/images/precision_sensitivity.svg
new file mode 100644
index 0000000..f6f4792
--- /dev/null
+++ b/materials/03-case_studies/images/precision_sensitivity.svg
@@ -0,0 +1,191 @@
+
+
+
+
diff --git a/materials/03-case_studies/images/southafrica_amplicon_heatmap.svg b/materials/03-case_studies/images/southafrica_amplicon_heatmap.svg
new file mode 100644
index 0000000..a22eb7a
--- /dev/null
+++ b/materials/03-case_studies/images/southafrica_amplicon_heatmap.svg
@@ -0,0 +1,1473 @@
+
+
+
+
diff --git a/materials/03-case_studies/images/southafrica_civet.png b/materials/03-case_studies/images/southafrica_civet.png
new file mode 100644
index 0000000..2a0d676
Binary files /dev/null and b/materials/03-case_studies/images/southafrica_civet.png differ
diff --git a/materials/03-case_studies/images/southafrica_multiqc_metrics.png b/materials/03-case_studies/images/southafrica_multiqc_metrics.png
new file mode 100644
index 0000000..7b8531c
Binary files /dev/null and b/materials/03-case_studies/images/southafrica_multiqc_metrics.png differ
diff --git a/materials/03-case_studies/images/southafrica_phylogeny.png b/materials/03-case_studies/images/southafrica_phylogeny.png
new file mode 100644
index 0000000..9091069
Binary files /dev/null and b/materials/03-case_studies/images/southafrica_phylogeny.png differ
diff --git a/materials/03-case_studies/images/switzerland_amplicon_heatmap.png b/materials/03-case_studies/images/switzerland_amplicon_heatmap.png
new file mode 100644
index 0000000..f44f293
Binary files /dev/null and b/materials/03-case_studies/images/switzerland_amplicon_heatmap.png differ
diff --git a/materials/03-case_studies/images/switzerland_multiqc_metrics.svg b/materials/03-case_studies/images/switzerland_multiqc_metrics.svg
new file mode 100644
index 0000000..82ab681
--- /dev/null
+++ b/materials/03-case_studies/images/switzerland_multiqc_metrics.svg
@@ -0,0 +1,3459 @@
+
+
+
+
diff --git a/materials/03-case_studies/images/switzerland_phylogeny.png b/materials/03-case_studies/images/switzerland_phylogeny.png
new file mode 100644
index 0000000..b5ee482
Binary files /dev/null and b/materials/03-case_studies/images/switzerland_phylogeny.png differ
diff --git a/materials/03-case_studies/images/switzerland_variants.svg b/materials/03-case_studies/images/switzerland_variants.svg
new file mode 100644
index 0000000..346e5d0
--- /dev/null
+++ b/materials/03-case_studies/images/switzerland_variants.svg
@@ -0,0 +1,2141 @@
+
+
+
+
diff --git a/materials/04-wastewater/00-ww_temp.html b/materials/04-wastewater/00-ww_temp.html
new file mode 100644
index 0000000..f7acb2d
--- /dev/null
+++ b/materials/04-wastewater/00-ww_temp.html
@@ -0,0 +1,629 @@
+
+
+
+
+
+
+
+
+
+SARS Genomic Surveillance
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
List some of the reasons why Linux is an essential operating system for bioinformatic analysis.
+
Distinguish between software containers and software environments.
+
Use package managers to install software globally and locally.
+
Recognise the advantage of using software containers for bioinformatic pipelines.
+
+
+
+
+
12.1 Why Linux?
+
Most computer users either use Windows or macOS as their operating system. However, in bioinformatics the Linux operating system is the most popular.
+
There are different reasons to prefer Linux, for example:
+
+
Most bioinformatics software is only available for this operating system.
+
Generally more secure.
+
Free and open source.
+
The operating system of choice on high performance compute (HPC) cluster environments.
+
It has flexible built-in command line tools to manipulate large data (cat, grep, sed, and the | pipe for chaining commands).
+
+
Because Linux is an open source project, there are many different types of Linux operating systems available. Generally, we recommend using Ubuntu, as it’s a well-supported and widely used distribution.
+
The first step for running bioinformatics software therefore is to ensure that you have access to Ubuntu Linux on your machine. We give instructions for this in Section 13.1.
+
+
+
12.2 Package Managers
+
Once you have Linux available, there is the question of how to install software, especially when using the command line. Most modern Linux operating systems have a default package manager available. A package manager allows the user to install (or remove, or upgrade) software with a single command. The package manager takes care of automatically downloading and installing the software we want, as well as any dependencies it requires.
+
+
+
+Diagram illustrating how package managers work. Image source.
+
+
+
+
12.2.1 Ubuntu apt
+
On Ubuntu, the default package manager is called apt. This software can be used to do system-wide updates, upgrades and installation of software packages. There are several commands available:
+
+
apt update updates the database of available packages, to check the latest versions available. This command doesn’t actually install anything.
+
apt upgrade upgrades all the packages to their latest version. This command upgrades existing packages, and installs new ones if required as dependencies.
+
apt install is used to install a single software of your choice.
+
+
For example, let’s say we wanted to install the latest version of java (Java is used for many applications, including IGV, which we introduced earlier). We could run the following commands:
+
sudo apt update # make sure the package database is up-to-date
+sudo apt upgrade # upgrade existing software to their latest version
+sudo apt install default-jre # install java
+
The sudo command at the beginning indicates that we want to run the apt command with administrator privilege, which will require you to input your password. This is necessary, because the apt command does a system installation of the software, so it can only be done if the user trying to do the installation has those permissions. With the install command, we give the name of the software we want to install. In this case, default-jre will install the default Java runtime environment.
+
The apt package manager is extremely useful to install core system software, however most bioinformatics software are not available from apt. Also, as it requires administrator (sudo) permissions, it is not always possible to install software with apt (for example, if you are using a HPC cluster). This is why we turn to alternative package managers such as Conda.
+
+
+
12.2.2 Debian Packages
+
Sometimes, software is not available through the apt repositories, but instead distributed as a file. Ubuntu uses the Debian package format. To install Debian packages, you can use the dpkg command.
+
For example, the RStudio application for Linux is distributed as a .deb file. After you download it, you can install it as follows:
+
sudo dpkg -i rstudio-2023.12.0-369-amd64.deb
+
As with apt this is a system installation and so requires admin privileges (sudo).
+
+
+
12.2.3Conda/Mamba Package Manager
+
Often you may want to use software packages that are not available on the apt repositories. A popular alternative in bioinformatics is to use the package manager Mamba, which is a successor to another package manager called Conda.
+
Conda and Mamba are package managers commonly used in data science, scientific computing, and bioinformatics. Conda, originally developed by Anaconda, is a package manager and environment manager that simplifies the creation, distribution, and management of software environments containing different packages and dependencies. It is known for its cross-platform compatibility and ease of use. Mamba is a more recent and high-performance alternative to Conda. While it maintains compatibility with Conda’s package and environment management capabilities, Mamba is designed for faster dependency resolution and installation, making it a better choice nowadays.
+
One of the strengths of using Mamba to manage your software is that you can have different versions of your software installed alongside each other, organised in environments. Organising software packages into environments is extremely useful, as it allows to have a reproducible set of software versions that you can use and reuse in your projects.
+
For example, imagine you are working on two projects with different software requirements:
+
+
Project A: requires Python 3.7, NumPy 1.15, and scikit-learn 0.20.
+
Project B: requires Python 3.9, the latest version of NumPy, and TensorFlow 2.0.
+
+
If you don’t use environments, you would need to install and maintain these packages globally on your system. This can lead to several issues:
+
+
Version conflicts: different projects may require different versions of the same library. For example, Project A might not be compatible with the latest NumPy, while Project B needs it.
+
Dependency chaos: as your projects grow, you might install numerous packages, and they could interfere with each other, causing unexpected errors or instability.
+
Difficulty collaborating: sharing your code with colleagues or collaborators becomes complex because they may have different versions of packages installed, leading to compatibility issues.
+
+
+
+
+Illustration of Conda/Mamba environments. Each environment is isolated from the others (effectively in its own folder), so different versions of the packages can be installed for distinct projects or parts of a long analysis pipeline.
+
+
+
Environments allow you to create isolated, self-contained environments for each project, addressing these issues:
+
+
Isolation: you can create a separate environment for each project using tools like Conda/Mamba. This ensures that the dependencies for one project don’t affect another.
+
Version control: you can specify the exact versions of libraries and packages required for each project within its environment. This eliminates version conflicts and ensures reproducibility.
+
Ease of collaboration: sharing your code and environment file makes it easy for collaborators to replicate your environment and run your project without worrying about conflicts.
+
Simplified maintenance: if you need to update a library for one project, it won’t impact others. You can manage environments separately, making maintenance more straightforward.
+
+
Another advantage of using Mamba is that the software is installed locally (by default in your home directory), without the need for admin (sudo) permissions.
+
You can search for available packages from the anaconda.org website. Packages are organised into “channels”, which represent communities that develop and maintain the installation “recipes” for each software. The most popular channels for bioinformatics and data analysis are “bioconda” and “conda-forge”.
+
There are three main commands to use with Mamba:
+
+
mamba create -n ENVIRONMENT-NAME: this command creates a new software environment, which can be named as you want. Usually people name their environments to either match the name of the main package they are installating there (e.g. an environment called pangolin if it’s to install the Pangolin software). Or, if you are installing several packages in the same environment, then you can name it as a topic (e.g. an environment called rnaseq if it contains several packages for RNA-seq data analysis).
+
mamba install -n ENVIRONMENT-NAME NAME-OF-PACKAGE: this command installs the desired package in the specified environment.
+
mamba activate ENVIRONMENT-NAME: this command “activates” the environment, which means the software installed there becomes available from the terminal.
+
+
Let’s see a concrete example. If we wanted to install packages for phylogenetic analysis, we could do:
+
# create an environment named "phylo"
+mamba create -n phylo
+
+# install some software in that environment
+mamba install -n phylo iqtree mafft
+
If we run the command:
+
mamba env list
+
We will get a list of environments we created, and “phylo” should be listed there. If we want to use the software we installed in that environment, then we can activate it:
+
mamba activate phylo
+
And usually this changes your terminal to have the word (phylo) at the start of your prompt.
+
+
+
+
12.3 Software Containers
+
Software containerization is a way to package software and its dependencies in a single file. A software container can be thought of as a very small virtual machine, with everything needed to run that software stored inside that file. For this reason, software containerization solutions, such as Docker and Singularity, are widely used in bioinformatics.
+
Software containers ensure reproducibility, allowing the same analysis to run on different systems. They can run on a local computer or on a high-performance computing cluster, producing the same result. The software within a container is isolated from other software, addressing the issue of incompatible dependencies between tools (similarly to Mamba environments).
+
As we already saw, analysis pipelines can be very complex, using many tools, each with their own dependencies. Therefore, worflow managers such as Nextflow use software containers to run their analysis (both Singularity and Docker are supported). This is what we’ve been doing throughout these materials, when running the nf-core/viralrecon pipeline, where we used the option -profile singularity. With this option, Nextflow will download the necessary software containers to run each step of the pipeline, which are available in public repositories online.
+
To use Singularity and Docker containers, the respective programs have to be installed, which we detail in our software setup page.
+
+
+
+
+
+
+Package managers or containers?
+
+
+
+
As we’ve seen, the Mamba package manager and the containerisation solutions such as Docker and Singularity are trying to achieve similar things: enabling the reproducible installation of software and its dependencies in an isolated environment. So, why use one or the other?
+
Mamba is more user-friendly, allowing you to easily install packages of your choice. However, Mamba often works less well for more complex environments and can become extremely inneficient for environments with too many packages and conflicting versions. The recommendation is to keep your Mamba environments relatively small and atomic (e.g. an environment for each software package or for small sets of related packages).
+
Singularity and Docker, on the other hand, allow for more complex environments. However, to create your own container requires more advanced knowledge and a steep learning curve. Fortunately, there are many existing containers available for bioinformatics, which Nextflow uses in its pipelines.
+
+
+
+
+
12.4 Summary
+
+
+
+
+
+
+Key Points
+
+
+
+
+
Linux is used in bioinformatics due to its open-source nature, powerful command-line interface, and compatibility with bioinformatics tools.
+
Package managers such as apt enable global software installations on Linux systems, simplifying the process of obtaining and managing software at the system level.
+
Local package managers such as mamba allow users to install and manage software within user-specific environments, avoiding conflicts with system-level packages.
+
Software containers, such as Docker and Singularity, encapsulate entire environments in a file and can be used to manage more complex software enviroments.
+
Bioinformatics workflow managers such as Nextflow can make use of software containers to run complex bioinformatic pipelines.
If you are attending one of our workshops, we will provide a virtual training environment with all of the required software and data pre-installed. If you want to setup your own computer to run the analysis demonstrated on this course, you can follow the instructions below.
+
+
+
+
+
+
+Hardware specifications
+
+
+
+
The viralrecon pipeline that we cover in this workshop can run on a regular desktop (e.g. with 4 CPUs and 16GB RAM). However, if you are processing hundreds of samples, it may take several hours or even days.
+
For general bioinformatic applications, we recommend investing in a high-performance desktop workstation. The exact specifications depend on the application, but as a minimum at least 32 threads and 64GB RAM.
The recommendation for bioinformatic analysis is to have a dedicated computer running a Linux distribution. The kind of distribution you choose is not critical, but we recommend Ubuntu if you are unsure.
Click the Windows key and search for Windows PowerShell, right-click on the app and choose Run as administrator.
+
Answer “Yes” when it asks if you want the App to make changes on your computer.
+
A terminal will open; run the command: wsl --install.
+Progress bars will show while installing “Virtual Machine Platform”, “Windows Subsystem for Linux” and finally “Ubuntu” (this process can take a long time).
+
+
Note: it has happened to us in the past that the terminal freezes at the step of installing “Ubuntu”. If it is frozen for ~1h at that step, press Ctrl + C and hopefully you will get a message saying “Ubuntu installed successfully”.
+
+
After installation completes, restart your computer.
+
After restart, a terminal window will open asking you to create a username and password.
+If it doesn’t, click the Windows key and search for Ubuntu, click on the App and it should open a new terminal.
+
+
You can use the same username and password that you have on Windows, or a different one - it’s your choice. Spaces and other special characters are not allowed for your Ubuntu username.
+
Note: when you type your password nothing seems to be happening as the cursor doesn’t move. However, the terminal is recording your password as you type. You will be asked to type the new password again to confirm it, so you can always try again if you get it wrong the first time.
+
+
+
You should now have access to a Ubuntu Linux terminal. This behaves very much like a regular Ubuntu server.
+
+
Configuring WSL2
+
After installation, it is useful to create shortcuts to your files on Windows. Your main C:\ drive is located in /mnt/c/ and other drives will be equally available based on their letter. To create shortcuts to commonly-used directories you use symbolic links. Here are some commands to automatically create shortcuts to your Windows “Documents”, “Desktop” and “Downloads” folders (copy/paste these commands on the terminal):
You may also want to configure the Windows terminal to automatically open WSL2 (instead of the default Windows Command Prompt or Powershell):
+
+
Search for and open the “ Terminal” application.
+
Click on the down arrow in the toolbar.
+
Click on “ Settings”.
+
Under “Default Profile” select “ Ubuntu”.
+
+
+
+
+
Another way to run Linux within Windows (or macOS) is to install a Virtual Machine. However, this is mostly suitable for practicing and not suitable for real data analysis.
+
Details for installing Ubuntu on VirtualBox is given on this page. Make sure to do these things, while you are setting it up:
+
+
In Step 2 “Create a user profile”: make sure to tick the Guest Additions option.
+
In Step 2 “Define the Virtual Machine’s resources”:
+
+
Assign at least 4 CPUs and 16000MB of RAM. At the very minimum you need 2 CPUs to run an Ubuntu VM.
+
Set at least 100GB as disk size, more if you have it available (note, this will not take 100GB of space on your computer, but it will allow using up to a maximum of that value, which is useful as we are working with sequencing data).
+
+
+
Once the installation completes, login to the Ubuntu Virtual machine, open a terminal and do the following:
+
+
Run su command.
+
Enter your user password. Your terminal should change to start with root@
+
Type the command: usermod -a -G sudo YOUR-USERNAME-HERE.
+
Close the terminal and restart the virtual machine.
+
+
These commands will add your newly created user to the “sudo” (admin) group.
+
+
+
+
After making a fresh install of Ubuntu, open a terminal and run the following commands to update your system and install some essential packages:
We recommend that you install Singularity and use the -profile singularity option when running Nextflow pipelines. On Ubuntu/WSL2, you can install Singularity using the following commands:
When using WSL2 on Windows, running Nextflow pipelines with -profile singularity sometimes doesn’t work.
+
As an alternative you can instead use Docker, which is another software containerisation solution. To set this up, you can follow the full instructions given on the Microsoft Documentation: Get started with Docker remote containers on WSL 2.
+
We briefly summarise the instructions here (but check that page for details and images):
This page lists some common file formats used in Bioinformatics (listed alphabetically). The heading of each file links to a page with more details about each format.
+
Generally, files can be classified into two categories: text files and binary files.
+
+
Text files can be opened with standard text editors, and manipulated using command-line tools (such as head, less, grep, cat, etc.). However, many of the standard files listed in this page can be opened with specific software that displays their content in a more user-friendly way. For example, the NEWICK format is used to store phylogenetic trees and, although it can be opened in a text editor, it is better used with a software such as FigTree to visualise the tree as a graph.
+
Binary files are often used to store data more efficiently. Typically, specific tools need to be used with those files. For example, the BAM format is used to store sequences aligned to a reference genome and can be manipulated with dedicated software such as samtools.
+
+
Very often, text files may be compressed to save storage space. A common compression format used in bioinformatics is gzip with has extension .gz. Many bioinformatic tools support compressed files. For example, FASTQ files (used to store NGS sequencing data) are often compressed with format .fq.gz.
Stores tabular data in a text file. (also see TSV format)
+
File extensions: .csv
+
+
These files can be opened with spreadsheet programs (such as Microsoft Excel). They can also be created from spreadsheet programs by going to File > Save As… and select “CSV (Comma delimited)” as the file format.
Stores tabular data in a text file. (also see CSV format)
+
File extensions: .tsv or .txt
+
+
These files can be opened with spreadsheet programs (such as Microsoft Excel). They can also be created from spreadsheet programs by going to File > Save As… and select “Text (Tab delimited)” as the file format.
+
+
+
+
+
\ No newline at end of file
diff --git a/materials/appendices/images/rstudio_data_viewer.svg b/materials/appendices/images/rstudio_data_viewer.svg
new file mode 100644
index 0000000..49e0e71
--- /dev/null
+++ b/materials/appendices/images/rstudio_data_viewer.svg
@@ -0,0 +1,1878 @@
+
+
+
+
diff --git a/materials/appendices/images/rstudio_panels.png b/materials/appendices/images/rstudio_panels.png
new file mode 100644
index 0000000..377612e
Binary files /dev/null and b/materials/appendices/images/rstudio_panels.png differ
diff --git a/materials/appendices/images/rstudio_setup.png b/materials/appendices/images/rstudio_setup.png
new file mode 100644
index 0000000..c55dae2
Binary files /dev/null and b/materials/appendices/images/rstudio_setup.png differ
diff --git a/materials/appendices/quick_r.html b/materials/appendices/quick_r.html
new file mode 100644
index 0000000..d4b1c3b
--- /dev/null
+++ b/materials/appendices/quick_r.html
@@ -0,0 +1,1450 @@
+
+
+
+
+
+
+
+
+
+SARS Genomic Surveillance
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
R fundamentals
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
This section gives a (very) quick and brief introduction to R and RStudio. It will not teach you the details of how to program in R, but it should serve as a basis to start you on that journey. R can be used for a range of data analysis applications, including statistics, machine learning, plotting, image analysis, bioinformatics, etc. R is also a programming language, which makes it very flexible. With this flexibility, comes a somewhat steeper learning curve, compared to point-and-click programs. However, in the long run it can really help you improve as a data analyst.
+
These materials will not teach you the details of how to write your own R programs (we won’t have time for these), but to make you familiar with some of its applications and how to use RStudio to run your code. It’s only the start of your journey into R programming.
+
If you want to learn more about this topic, we recommend:
R is the software and programming language itself, but the R interface is quite basic and not very user-friendly. Fortunately, there is a companion piece of software called RStudio, which makes working with R a little bit easier.
+
There are 4 basic panels in RStudio (see image below):
+
+
The script panel is essentially a text editor, where we can write code and save it as a text file, which in this case, because it contains R code, we call it an R script (but remember, a script is just a text file with some code in it. We’ve been creating shell scripts that run on the command line, here we have some R scripts, which contain R code)
+
The console panel is where the code actually runs, or is executed. This is equivalent to the terminal, on the command line. If we want to execute a line of code, then we need to run it on the console.
+
+
One nice feature of RStudio is that we can edit our code on the script panel and then run a line of code from the script on the console - it’s like copy/pasting that line of code from the script to the console. This makes working interactively with RStudio much easier, because we can edit our code in the script and run it as we go along.
+
+
On the top-right we have the Environment panel, which shows us objects that we create, that store information such as tables of data that we read into R.
+
Finally, on the bottom-right there are a few tabs: a file browser (allowing us to see files in our computer), a plot display tab (for plots we generate) and a help tab to look at documentation.
+
+
+
+
Setting RStudio
+
Before we start working with RStudio, it’s a good idea to change one of its default options. Go to Tools → Global Options… and change the following:
+
+
This will tell RStudio to NOT automatically save and load things that we have done in the past. You may think this is a helpful thing, but actually it’s very inconvenient, because if you are doing multiple analysis, it might get very very confusing what the objects that you created are! So, it’s always best to start R with a fresh session, and setting these options makes sure we do this.
+
+
+
Starting a Project
+
R has a concept called working directory, which is the location on your computer where it is working from (looking for files and folders). You can think of it as the folder that you cd into if you were working on the command-line.
+
The easiest way to ensure that R is using the correct working directory for our analysis, is to create an R project. In RStudio: File → New Project… → Existing Directory and then click the Browse… button to navigate to the folder where your project files are located.
+
This will create an .Rproj file on your project folder. Next time you want to work on your analysis, you can simply double-click on this file, and it will open RStudio, with the correct working directory already set for you.
+
+
+
+
R Basics
+
This section introduces some of the basic concepts in the R programming language.
+
+
Packages/Libraries
+
R has several extensions to the basic functionality called “packages” or “libraries”. A popular library for data manipulation is called tidyverse, which we are using in this course. Each time we start a new R session, we need to load the libraries we want to use:
+
+
library(tidyverse)
+
+
If you get an error like Error in library(tidyverse) : there is no package called 'tidyverse', that means that you didn’t install the package. To install packages you can run:
+
+
install.packages("tidyverse")
+
+
You only need to do this the first time you want to use a library. Once it’s installed, you don’t need to run this command again (unless you want to update the library to its latest version – often a good idea!).
+
+
+
Create objects
+
Create objects (something that contains a value) with <-. For example, the following creates an object called x containing a single number:
+
+
x <-53.341
+
+
We can print the content of the object by typing its name:
+
+
x
+
+
[1] 53.341
+
+
+
+
+
Functions
+
Most of the tasks we can achieve in R are done through the use of functions. We can think of functions as mini-programs that take an input and give an output.
+
Functions are easy to identify, because they are always followed by parenthesis. Inside the parenthesis we include the inputs to the function.
+
+
round(x) # round the the value of x
+
+
[1] 53
+
+
+
Functions have options that can change their behaviour. Separate options using a comma:
+
+
round(x, digits =1) # round to one decimal point
+
+
[1] 53.3
+
+
+
+
+
Vector
+
A vector is the most basic type of object in R. It is a collection of values, which are all of the same type, for example numeric, character or logical (TRUE/FALSE).
In many situations (for example to filter rows in a table), it’s useful to evaluate a set of conditions. We can create logical vectors using conditions:
+
+
x_num
+
+
[1] 1.0 5.0 23.3 55.2
+
+
# is x_num greater than 20?
+x_num >20
+
+
[1] FALSE FALSE TRUE TRUE
+
+
# is x_num equal to 5?
+x_num ==5
+
+
[1] FALSE TRUE FALSE FALSE
+
+
# is x_num contained the vector on the right?
+x_num %in%c(20, 30, 1)
+
+
[1] TRUE FALSE FALSE FALSE
+
+
+
Combine conditions with & (AND) and | (OR):
+
+
x_num
+
+
[1] 1.0 5.0 23.3 55.2
+
+
# is x_num greater than or equal to 10 AND smaller than or equal to 30?
+x_num >=10& x_num <=30
+
+
[1] FALSE FALSE TRUE FALSE
+
+
# is x_num smaller than 10 OR greater than 30?
+x_num <10| x_num >30
+
+
[1] TRUE TRUE FALSE TRUE
+
+
+
To set the filtering conditions, several relational operators can be used:
+
+
== is equal to
+
!= is different from
+
%in% is contained in
+
> is greater than
+
>= is greater than or equal to
+
< is less than
+
<= is less than or equal to
+
+
It is also possible to combine several conditions together using the following logical operators:
+
+
& AND
+
| OR
+
+
+
+
Missing Values
+
Sometimes we have missing values in our data, which are encoded as NA:
+
+
y <-c(23, 44, NA, 212)
+
+
We need to ensure these are dealt with properly
+
+
# returns NA
+mean(y)
+
+
[1] NA
+
+
# removes NA and then calculates mean
+mean(y, na.rm =TRUE)
+
+
[1] 93
+
+
+
The is.na() function is important to deal with missing values:
+
+
y
+
+
[1] 23 44 NA 212
+
+
# create a logical that is true if value is missing
+is.na(y)
+
+
[1] FALSE FALSE TRUE FALSE
+
+
# Negate that expression using !
+!is.na(y)
+
+
[1] TRUE TRUE FALSE TRUE
+
+
+
+
+
Tables: data.frame/tibble
+
Tables in R are called data.frame. The tidyverse package has its own version of a data.frame called a tibble. For the most part they are basically equivalent, but the tibble object has a nicer printing function to display our data on the console.
+
As an example for working with tables in R, let’s read a TSV (tab-delimited) file that contains intervals of missing information in 5 SARS-CoV-2 consensus sequences (this data comes from the Switzerland case study). To import a TSV file into R as a data.frame we can use the function read_tsv() (for a CSV file we would use read_csv()):
Rows: 132 Columns: 6
+── Column specification ────────────────────────────────────────────────────────
+Delimiter: "\t"
+chr (4): seqID, patternName, pattern, strand
+dbl (2): start, end
+
+ℹ Use `spec()` to retrieve the full column specification for this data.
+ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
+
+
+
When we read the table in, we get a message informing us of the column types found. In this case we have character columns containing text (indicated by chr) and numeric columns (indicated by dbl, which refers to the double-precission floating point format that computers use to store numbers).
+
To see the content of the table you can type the name of the object:
Sometimes a more convenient way is to click the name of the table on the environment, which opens a new tab to preview your data.
+
+
+
+Data viewer in RStudio.
+
+
+
+
+
+
Data Manipulation
+
Most of the work you will do in R is with tables of data (data.frame/tibble objects). There are several ways to manipulate tables in R, but we will give a quick overview of the functionality available through the tidyverse collection of packages.
+
+
Basic “verbs”
+
There’s a set of basic functions that can be thought of as “data manipulation verbs”. They are:
+
+
mutate() → add a new column of modify an existing one.
+
select() → select columns from the table.
+
filter() → subset the rows from the table that fullfill a certain logical condition.
+
+
Here are some examples of each:
+
+
# create a new column with the missing interval lengths
+mutate(missing_intervals,
+length = (end - start) +1)
We can chain multiple commands together using pipes (similarly to pipes in Unix). In R the pipe is represented by |> (or %>%). The way the pipe works is that the output of one function is sent as the input to the next function.
+
Taking the examples from the previous section, we could chain all those commands like this:
We can calculate summaries of the data (e.g. mean, standard deviation, maximum, minimum) per group (e.g. per sample) using a pair of functions together. For example:
+
+
# mean and maximum interval length per sample
+missing_intervals |>
+# for each sample
+group_by(seqID) |>
+# calculate summary statistics
+summarise(max_length =max(length),
+min_length =min(length),
+mean_length =mean(length))
As before, if we wanted to save this result in a new object, we would need to use <-:
+
+
intervals_summary <- missing_intervals |>
+# for each sample
+group_by(seqID) |>
+# calculate summary statistics
+summarise(max_length =max(length),
+min_length =min(length),
+mean_length =mean(length)) |>
+# and rename the seqID column
+rename(sample = seqID)
+
+
Notice in this case we also renamed the column called seqID to be named sample instead (this will be useful for the exercise later on).
+
Another useful function is count(), which counts how many times the values in a column appear on a data frame. For example, if we wanted to know how many missing intervals each sample had, we could do it like this:
We can join multiple tables together based on a common identifier. There are different types of join operations, depending on what we want to achieve.
+
Take these two tables as an example (these tables come pre-loaded with tidyverse):
+
+
band_members
+
+
# A tibble: 3 × 2
+ name band
+ <chr> <chr>
+1 Mick Stones
+2 John Beatles
+3 Paul Beatles
+
+
band_instruments
+
+
# A tibble: 3 × 2
+ name plays
+ <chr> <chr>
+1 John guitar
+2 Paul bass
+3 Keith guitar
+
+
+
Here are some different ways we can join these tables:
+
+
# keep all records from both tables
+full_join(band_members, band_instruments, by ="name")
+
+
# A tibble: 4 × 3
+ name band plays
+ <chr> <chr> <chr>
+1 Mick Stones <NA>
+2 John Beatles guitar
+3 Paul Beatles bass
+4 Keith <NA> guitar
+
+
# keep all records from the first table
+left_join(band_members, band_instruments, by ="name")
+
+
# A tibble: 3 × 3
+ name band plays
+ <chr> <chr> <chr>
+1 Mick Stones <NA>
+2 John Beatles guitar
+3 Paul Beatles bass
+
+
# keep all records from the second table
+right_join(band_members, band_instruments, by ="name")
+
+
# A tibble: 3 × 3
+ name band plays
+ <chr> <chr> <chr>
+1 John Beatles guitar
+2 Paul Beatles bass
+3 Keith <NA> guitar
+
+
# keep only the records occurring in both tables
+inner_join(band_members, band_instruments, by ="name")
+
+
# A tibble: 2 × 3
+ name band plays
+ <chr> <chr> <chr>
+1 John Beatles guitar
+2 Paul Beatles bass
+
+
+
In each case, if there was no match between the two tables, the cells are filled with missing values NA.
+
+
+
+
+
+
+Exercise
+
+
+
+
+
+
+
+
In this exercise we will join the intervals_summary table we created earlier, with the metadata table that contains information about our samples.
+
+
Read the your metadata table sample_info.csv into R and save it as an object called sample_info.
+
+
+Hint
+
+Remember that you can use the read_csv() function to read CSV files into R.
+
+
Join the sample_info table that you just imported with the intervals_summary table we created earlier. Save the output to the intervals_summary table (this will update the table).
+
+
+You can use the left_join() function to achieve this, using the “sample” column as the identifier column used to join the two tables.
+
+
+
+
+
+
+
+
+
+Answer
+
+
+
+
+
+
+
+
Answer 1
+
To read the metadata CSV file, we use the read_csv() function, and use <- to save the output in an object called sample_info:
+
+
sample_info <-read_csv("sample_info.csv")
+
+
Rows: 7 Columns: 4
+── Column specification ────────────────────────────────────────────────────────
+Delimiter: ","
+chr (3): sample, country, sequencing_instrument
+date (1): collection_date
+
+ℹ Use `spec()` to retrieve the full column specification for this data.
+ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
+
+
+
We can see the content of our file by typing its name in the console. Here is the file in our example data (yours might look different):
To join the two tables together, we can use one of the *_join() functions. In this case it doesn’t matter which function we use, because both tables have the same sample IDs. But, for example, let’s say we only wanted to retain the samples that are in common across both tables. In that case, we would use inner_join():
+
+
intervals_summary <-inner_join(intervals_summary, sample_info, by ="sample")
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Data Visualisation
+
For this section, we will use another table, which contains some of the metrics that we can collect from our consensus pipeline:
+
+
# read the table
+qc_metrics <-read_tsv("consensus_metrics.tsv")
+
+
Rows: 7 Columns: 6
+── Column specification ────────────────────────────────────────────────────────
+Delimiter: "\t"
+chr (3): sample, qc_status, lineage
+dbl (3): n_mapped_reads, median_depth, pct_missing
+
+ℹ Use `spec()` to retrieve the full column specification for this data.
+ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
+
+
# preview the table
+qc_metrics
+
+
# A tibble: 7 × 6
+ sample n_mapped_reads median_depth pct_missing qc_status lineage
+ <chr> <dbl> <dbl> <dbl> <chr> <chr>
+1 CH01 43900 248 3.05 good BA.1
+2 CH02 42841 159 4.20 good BA.1
+3 CH03 40079 160 3.77 good BA.1
+4 CH04 50902 164 12.5 bad BA.1
+5 CH05 32020 133 10.0 bad BA.1
+6 CH06 46277 177 8.87 mediocre BA.1.1
+7 CH07 15867 41 33.2 bad None
+
+
+
We can build plots from our tables using the ggplot2 package (which is also part of the tidyverse).
+
To build a ggplot, we usually need at least three things:
+
+
The data frame we want to use for the plot (the data).
+
The columns we want to visualise as our x-axis, y-axis, and colours (these are called aesthetics).
+
The type of shape that we want to plot (these are called geometries).
+
+
For example, let’s try to make a plot to show the relationship between total number of counts and the median depth of sequencing in these samples:
+
+
ggplot(data = qc_metrics, aes(x = n_mapped_reads, y = median_depth))
+
+
+
+
+
When we do this, we simply get an empty plot, with x and y axis, but nothing drawn on it. To draw something on the plot, we add (literally with +) geometries to our plot. In this case, we can use the geom_point() geometry, which draws “points” on the plot:
geom_col() draws a barplot (this one is named a little strangely, but “col” means it draws “columns” or bars).
+
+
We can further modify the look to the plot by adding other aesthetics such as the colour of the points. For example, let’s say we wanted to colour our points according to their “QC Status”:
Modify the plot we just did, to show the relationship between median depth of sequencing (x-axis) and percentage of missing bases (y-axis). Colour the points according to the lineage column.
Primers for the “Midnight” protocol, also known as “1200” as it produces fragments that are ~1200bp long. This primer scheme is optimised for ONT platforms (link).
+
To analyse with nf-core/viralrecon, add these options to the command:
The “xGen SARS-CoV-2 Amplicon Panels”, previously called “SWIFT Normalase”, is commercialised by IDT. Unfortunately IDT does not make their BED files available publicly. If you purchase a kit from IDT, the company should provide you with the correct BED file to process your samples.
+
NOTE: in the future we will provide a link to a BED file with approximate coordinates, inferred bioinformatically. This can be useful if re-analysing public data.
+
+
+
+
Data Resources
+
+
outbreak.info - interactive exploration of global SARS-CoV-2 data.
This document gives a brief summary of useful Unix commands. Anything within {} indicates a user-provided input.
+
+
+
+
+
+
+Note
+
+
+
+
If you are on Windows, you can install the Windows Subsystem for Linux following our instructions.
+
+
+
+
Documentation and Help
+
+
+
+
+
+
+
+
man {command}
+
manual page for the program
+
+
+
whatis {command}
+
short description of the program
+
+
+
{command} --help
+
many programs use the --help flag to print documentation
+
+
+
+
+
+
Listing files
+
+
+
+
ls
+
list files in the current directory
+
+
+
ls {path}
+
list files in the specified path
+
+
+
ls -l {path}
+
list files in long format (more information)
+
+
+
ls -a {path}
+
list all files (including hidden files)
+
+
+
+
+
+
Change Directories
+
+
+
+
+
+
+
+
cd {path}
+
change to the specified directory
+
+
+
cd or cd ~
+
change to the home directory
+
+
+
cd ..
+
move back one directory
+
+
+
pwd
+
print working directory. Shows the full path of where you are at the moment (useful if you are lost)
+
+
+
+
+
+
Make or Remove Directories
+
+
+
+
+
+
+
+
mkdir {dirname}
+
create a directory with specified name
+
+
+
rmdir {dirname}
+
remove a directory (only works if the directory is empty)
+
+
+
rm -r {dirname}
+
remove the directory and all it’s contents (use with care)
+
+
+
+
+
+
Copy, Move and Remove Files
+
+
+
+
+
+
+
+
cp {source/path/file1} {target/path/}
+
copy “file1” to another directory keeping its name
+
+
+
cp {source/path/file1} {target/path/file2}
+
copy “file1” to another directory naming it “file2”
+
+
+
cp {file1} {file2}
+
make a copy of “file1” in the same directory with a new name “file2”
+
+
+
mv {source/path/file1} {target/path/}
+
move “file1” to another directory keeping its name
+
+
+
mv {source/path/file1} {target/path/file2}
+
move “file1” to another directory renaming it as “file2”
+
+
+
mv {file1} {file2}
+
is equivalent to renaming a file
+
+
+
rm {filename}
+
remove a file
+
+
+
+
+
+
View Text Files
+
+
+
+
+
+
+
+
less {file}
+
view and scroll through a text file
+
+
+
head {file}
+
print the first 10 lines of a file
+
+
+
head -n {N} {file}
+
print the first N lines of a file
+
+
+
tail {file}
+
print the last 10 lines of a file
+
+
+
tail -n {N} {file}
+
print the last N lines of a file
+
+
+
head -n {N} {file} | tail -n 1
+
print the Nth line of a file
+
+
+
cat {file}
+
print the whole content of the file
+
+
+
cat {file1} {file2} {...} {fileN}
+
concatenate files and print the result
+
+
+
zcat {file} and zless {file}
+
like cat and less but for compressed files (.zip or .gz)
+
+
+
+
+
+
Find Patterns
+
Finding (and replacing) patterns in text is a very powerful feature of several command line programs. The patterns are specified using regular expressions (shortened as regex), which are not covered in this document. See this Regular Expressions Cheat Sheet for a comprehensive overview.
+
+
+
+
+
+
+
+
grep {regex} {file}
+
print the lines of the file that have a match with the regular expression pattern
+
+
+
+
+
+
Wildcards
+
+
+
+
+
+
+
+
*
+
match any number of characters
+
+
+
?
+
match any character only once
+
+
+
Examples
+
+
+
+
ls sample*
+
list all files that start with the word “sample”
+
+
+
ls *.txt
+
list all the files with .txt extension
+
+
+
cp * {another/directory}
+
copy all the files in the current directory to a different directory
+
+
+
+
+
+
Redirect Output
+
+
+
+
+
+
+
+
{command} > {file}
+
redirect output to a file (overwrites if the file exists)
+
+
+
{command} >> {file}
+
append output to a file (creates a new file if it does not already exist)
+
+
+
+
+
+
Combining Commands with | Pipes
+
+
+
+
+
+
+
+
<command1> | <command2>
+
the output of “command1” is passed as input to “command2”
+
+
+
Examples
+
+
+
+
ls | wc -l
+
count the number of files in a directory
+
+
+
cat {file1} {file2} | less
+
concatenate files and view them with less
+
+
+
cat {file} | grep "{pattern}" | wc -l
+
count how many lines in the file have a match with “pattern”
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
\ No newline at end of file
diff --git a/search.json b/search.json
new file mode 100644
index 0000000..b01bdfa
--- /dev/null
+++ b/search.json
@@ -0,0 +1,758 @@
+[
+ {
+ "objectID": "index.html",
+ "href": "index.html",
+ "title": "Overview",
+ "section": "",
+ "text": "Overview\nThese materials provide a practical guide on analysing viral amplicon sequencing data for genomic surveillance, with a specific focus on SARS-CoV-2. While centered on SARS-CoV-2, the concepts and pipelines explored here are applicable to various viruses. The content includes the analysis of data from clinical isolates and wastewater samples. For clinical isolates, we illustrate how to create consensus sequences for upload to databases like GISAID and for downstream applications such as variant annotation and phylogeny. Wastewater sample analysis includes estimating variant and mutation frequencies. For both applications we will use a standardized bioinformatic pipeline compatible with both Illumina and Nanopore data. The materials cover assigning sequences to lineages, identifying variants of interest and creating visualizations to effectively communicate findings. Throughout, you will acquire foundational bioinformatic skills, including Unix command line usage and scripting for reproducible analyses."
+ },
+ {
+ "objectID": "index.html#citation",
+ "href": "index.html#citation",
+ "title": "Overview",
+ "section": "Citation",
+ "text": "Citation\n\nPlease cite these materials if:\n\nYou adapted or used any of them in your own teaching.\nThese materials were useful for your research work. For example, you can cite us in the methods section of your paper: “We carried our analyses based on the recommendations in Tavares et al (2022).”.\n\nYou can reference these materials as:\n\nTavares H, Salehe B, Kumar A, Castle M & UKHSA New Variant Assessment Platform Team (2022) “cambiotraining/sars-cov-2-genomics: Introduction to Sars-CoV-2 Genomics”, https://cambiotraining.github.io/sars-cov-2-genomics\n\nOr, in BibTeX format:\n@Misc{,\n author = {Tavares, Hugo and Salehe, Bajuna and Kumar, Ankit and Castle, Matt and UKHSA New Variant Assessment Platform Team},\n title = {cambiotraining/sars-cov-2-genomics: Introduction to Sars-CoV-2 Genomics},\n month = {March},\n year = {2022},\n url = {https://cambiotraining.github.io/sars-cov-2-genomics},\n}\nPlease make sure to include a link to the materials in the citation. (we will add a DOI in due time)\nThe contributing members from University of Cambridge Bioinformatics Training Facility team are:\n\nMatt Castle, Bioinformatics Training Manager\nHugo Tavares, Senior Teaching Associate\nBajuna Salehe, Teaching Associate\nAnkit Kumar, Teaching Assistant\n\nThe UKHSA’s NVAP Team members that supported these materials are:\n\nDr Leena Inamdar, NVAP Programme Lead and Global Health Lead\nDr Babak Afrough, Senior Project Manager\nAude Wilhelm, Senior Epidemiology Scientist\nRichard Myers, Data Analytics Surveillance Head Bioinformatician\nSam Sims, Bioinformatician\nKate Edington, Bioinformatician\nConstantina Laou, Specialist Lab Advisor"
+ },
+ {
+ "objectID": "index.html#acknowledgements",
+ "href": "index.html#acknowledgements",
+ "title": "Overview",
+ "section": "Acknowledgements",
+ "text": "Acknowledgements\nThese materials have been developed as a collaboration between the Bioinformatics Training Facility at the University of Cambridge and the New Variant Assessment Platform (NVAP) program from the UK Health Security Agency.\nOur partners also include COG Train. We also thank the wider community for publicly sharing training resources, including:\n\nThe workshop video series from CLIMB BIG DATA.\nThe Carpentries project, in particular for their Unix Shell lesson, which we adapted for this workshop."
+ },
+ {
+ "objectID": "setup.html#software",
+ "href": "setup.html#software",
+ "title": "Data & Software",
+ "section": "Software",
+ "text": "Software\nThe software installation for this course is quite complex and detailed in a separate section."
+ },
+ {
+ "objectID": "setup.html#data",
+ "href": "setup.html#data",
+ "title": "Data & Software",
+ "section": "Data",
+ "text": "Data\nDifferent datasets are used throughout these materials. This page provides links to download each dataset, with a brief description for each of them. We have split the data between sequencing platforms."
+ },
+ {
+ "objectID": "setup.html#illumina",
+ "href": "setup.html#illumina",
+ "title": "Data & Software",
+ "section": "Illumina",
+ "text": "Illumina\n\nUK Sequences\nThese samples were downloaded from SRA and include samples collected in the UK. These data are used in the following sections of the materials: Consensus Assembly, Lineages and Variants and Building Phylogenies.\n\n Download (zip file)\n\n\n\nClick here to see details of how the data was downloaded from public repositories\n\nWe obtained these data from the SRA repository, using the fastq-dump command as follows:\nfastq-dump --split-3 --gzip ERR5728910 ERR5728911 ERR5728913 ERR5742126 ERR5742297 ERR5742457 ERR5742549 ERR5742553 ERR5761182 ERR5761193 ERR5765358 ERR5770082 ERR5855061 ERR5855065 ERR5855555 ERR5914874 ERR5921129 ERR5921248 ERR5921268 ERR5921355 ERR5921612 ERR5925864 ERR5926784 ERR5932087 ERR5932097 ERR5932290 ERR5932412 ERR5932418 ERR5932680 ERR5932985 ERR5933082 ERR5933090 ERR6106244 ERR6083647 ERR6085882 ERR6086247 ERR6104816 ERR6105221 ERR6105244 ERR6105337 ERR6105341 ERR6106514 ERR6106801 ERR6107074 ERR6128896 ERR6128978 ERR6129122 ERR6129126\n\n\n\n\nSouth Africa\nThese samples were downloaded from SRA and include samples collected between Nov and Dec 2021 in South Africa. These data are used in the worked example South Africa (Illumina).\nWe provide two versions of the data: the full data includes 24 samples (more realistic sample size), whereas the small version includes only 8 samples (quicker processing time for practising).\n\n Full data - 24 samples (zip file)\n Small data - 8 samples (zip file)\n\n\n\nClick here to see details of how the data was downloaded from public repositories\n\nWe obtained these data from the SRA repository, using the fastq-dump command as follows:\nfastq-dump --split-3 --gzip SRR17051908 SRR17051923 SRR17051916 SRR17051953 SRR17051951 SRR17051935 SRR17051932 SRR17054503 SRR17088930 SRR17088928 SRR17088924 SRR17088917 SRR17461712 SRR17461700 SRR17712997 SRR17712994 SRR17712779 SRR17701841 SRR17712711 SRR17701832 SRR17701890 SRR17712607 SRR17712594 SRR17712442 SRR17712435 SRR17712343 SRR17712341 SRR17712321 SRR17712313 SRR17712312 SRR17712387 SRR17970983 SRR17973983 SRR17973948 SRR17973937 SRR17973974 SRR17974004 SRR17974001 SRR17974000 SRR17973999 SRR17973998 SRR17973997 SRR17973996 SRR17973995 SRR17973992 SRR17973991 SRR17973989 SRR17973988\n\n\n\n\nEQA panels\nThe standard panels for External Quality Assessment provided by NEQAS were sequenced on an Illumina platform by UKHSA and used to generate four sets of data for training purposes. These data can be used for the extended exercise: EQA (Exercise).\nWe provide a link to each of the four datasets below (the datasets are very similar to each other):\n\n Dataset 1 (zip file)\n Dataset 2 (zip file)\n Dataset 3 (zip file)\n Dataset 4 (zip file)"
+ },
+ {
+ "objectID": "setup.html#nanopore",
+ "href": "setup.html#nanopore",
+ "title": "Data & Software",
+ "section": "Nanopore",
+ "text": "Nanopore\n\nIndia\nThese samples were downloaded from SRA and include samples collected in India and sequenced by the National Institute of Mental Health and Neurosciences. These data are used for the exercises in the following sections of the materials: Consensus Assembly, Lineages and Variants and Building Phylogenies.\n\n Download (zip file)\n\nPlatform: MinION\nNanopore chemistry: not specified (we assumed 9.4.1)\nPrimer set: not specified (we assumed ARTIC primers v3)\nGuppy version: not specified (we assumed 3.6 or higher)\nBasecalling mode: not specified (we assumed “high”)\n\n\nNote that several details needed to run the medaka pipeline were not provided in the public repository. We arbitrarily picked parameters as detailed above, but this is for demonstration purposes only.\n\n\nClick here to see details of how the data was downloaded from public repositories\n\nWe obtained these data from the SRA repository, using the fastq-dump command as follows:\nfastq-dump --split-3 --gzip SRR14494107 SRR14493634 SRR14493632 SRR14493631 SRR14493707 SRR14493705 SRR14493730 SRR14493729 SRR14493728 SRR14493727 SRR14493726 SRR14493725 SRR14493724 SRR14493723 SRR14493722 SRR14493721 SRR14493719 SRR14493718 SRR14493717 SRR14493716 SRR14493715 SRR14493714 SRR14493713 SRR14493712 SRR14493711 SRR14494106 SRR14494095 SRR14494092 SRR14494091 SRR14494090 SRR14494089 SRR14494088 SRR14494087 SRR14494086 SRR14494105 SRR14494104 SRR14494103 SRR14494102 SRR14494101 SRR14494100 SRR14493626 SRR14494099 SRR14494098 SRR14494097 SRR14494096 SRR14494094 SRR14494093\n\n\n\n\nSwitzerland\nThese samples were downloaded from SRA and include samples collected between Nov 2021 and Jan 2022 in Switzerland and sequenced by the Institute for Infectious Diseases, University of Bern. These data are used in the worked example Switzerland (Nanopore).\nWe provide two versions of the data: the full data includes 65 samples (more realistic sample size), whereas the small version includes only 10 samples (quicker processing time for practising).\n\n Full data - 65 samples (zip file)\n Small data - 10 samples (zip file)\n\nPlatform: GridION\nNanopore chemistry: not specified (we assumed 9.4.1)\nPrimer set: ARTIC primers v3\nGuppy version: not specified (we assumed 3.6 or higher)\nBasecalling mode: not specified (we assumed “fast”)\n\n\nNote that several details needed to run the medaka pipeline were not provided in the public repository. We arbitrarily picked parameters as detailed above, but this is for demonstration purposes only.\n\n\nClick here to see details of how the data was downloaded from public repositories\n\nWe obtained these data from the SRA repository, using the fastq-dump command as follows:\nfastq-dump --split-3 --gzip ERR8971298 ERR8961150 ERR8961147 ERR8961133 ERR8961130 ERR8961129 ERR8961128 ERR8961124 ERR8961123 ERR8961116 ERR8961115 ERR8961114 ERR8961112 ERR8961110 ERR8961065 ERR8961062 ERR8961333 ERR8959962 ERR8959961 ERR8959960 ERR8959959 ERR8959958 ERR8959957 ERR8959956 ERR8959955 ERR8959953 ERR8959952 ERR8959950 ERR8959949 ERR8959948 ERR8959946 ERR8959945 ERR8959943 ERR8959942 ERR8959940 ERR8959939 ERR8959938 ERR8959937 ERR8959936 ERR8959934 ERR8959933 ERR8959931 ERR8959927 ERR8959926 ERR8959925 ERR8959912 ERR8959911 ERR8959905 ERR8959901 ERR8959892 ERR8960229 ERR8960215 ERR8960216 ERR8960217 ERR8960218 ERR8960219 ERR8960220 ERR8960221 ERR8960223 ERR8959343 ERR8959341 ERR8959330 ERR8959327\n\n\n\n\nEQA panels\nThe standard panels for External Quality Assessment provided by NEQAS were sequenced on an ONT platform by TODO. These data can be used for the extended exercise: EQA (Exercise).\nWe provide a link to datasets corresponding to two different runs:\n\n Dataset 1 (zip file)\n\nPlatform: PromethION\nNanopore chemistry: 9.4.1\nPrimer set: midnight\nGuppy version: 6.6\nBasecalling mode: “fast”\n\n Dataset 2 (zip file)\n\nPlatform: GridION\nNanopore chemistry: 9.4.1\nPrimer set: 4.1\nGuppy version: 6.6\nBasecalling mode: “high”\n\n\nWe thank the collaborating institutions for sharing their trainining data with us."
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#what-is-sars-cov-2",
+ "href": "materials/01-intro/01-surveillance.html#what-is-sars-cov-2",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.1 What is SARS-CoV-2?",
+ "text": "1.1 What is SARS-CoV-2?\nSARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2) is a betacoronavirus resposible for the COVID-19 disease and caused a global outbreak leading to an ongoing pandemic. The initial spread of this virus started in the city of Wuhan, in China. Despite early efforts to contain its spread in China (through several lockdowns in the country), the virus spread to other provinces within China and, eventually, to other countries across the world. This led to the World Health Organisation (WHO) declaring a public health emergency on 30 January 2020 and then a pandemic on 11 March 2020.\nSARS-CoV-2 is an RNA virus, composed of single-stranded RNA. The first SARS-CoV-2 genome was published in January 2020 and is approximately 30Kb long. It encodes several proteins including the so-called Spike protein (or ‘S’ for short), which is used by the virus to interact and eventually enter human cells and cause infection. This interaction happens by the binding of the S protein to the ACE2 protein receptor found in human cells.\n\n\n\nGenome structure of SARS-CoV-2. Source: Rahimi, Mirzazadeh & Tavakolpour 2021\n\n\nThe genome sequence of SARS-CoV-2 has been a huge contributor to our ability to manage current pandemic. Namely:\n\nIt allowed the development of a vaccine to target the S protein.\nIt allowed the development of diagnostic tests for positive cases (lateral flow test and PCR-based test).\nIt allowed the development of protocols for whole-genome sequencing of the virus from positive human samples.\n\nThis last point is the focus of our workshop, and we will spend some time looking at how to analyse these data.\nYou can watch the video below, created by the Maastrich University, if you want to learn more about the lifecycle of the SARS-CoV-2 virus and the role that its different proteins play in this context:"
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#genomic-surveillance",
+ "href": "materials/01-intro/01-surveillance.html#genomic-surveillance",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.2 Genomic Surveillance",
+ "text": "1.2 Genomic Surveillance\nThe number of SARS-CoV-2 genomes sequenced is the largest of a pathogen ever done. As such, it has enabled us to track how the virus evolves and spreads both at a local and global scale at an unprecendented resolution. This allowed the identification of mutations that affect characteristics of the virus, such as its transmissibility or severity of the disease, and testing new strains of the virus to understand whether they are effectively neutralised by current or future vaccines.\nOne of the main applications of SARS-CoV-2 sequencing is to infer relationships between the different circulating forms of the virus. This is done by comparing these sequences and building phylogenetic trees that reflect their sequence similarities. From these, it is possible to identify particular clusters of similar sequences that spread faster than expected, and may therefore be associated with mutations that increase the virus’ fitness.\n\n\n\nExample of global phylogeny from the Nextstrain public server. Colours show clusters of similar sequences. (Screenshot taken Feb 2022)\n\n\nFor example, one of the first mutations identified from these sequencing efforts was A23403G (an adenine was replaced by a guanine in position 23,403 of the genome), which caused an aminoacid substitution in the S protein that increased virus infectivity and transmissibility. Since then, many new forms of the virus have been identified as being of particular concern for public health, and have been classified by the WHO as Variants of Concern. These have been named based on letters of the Greek alphabet, and have been a crucial way to inform public health policy and containment measures around the world.\nIn addition to the WHO variant nomenclature, there are three main projects that have been instrumental in grouping SARS-CoV-2 sequences into similar groups:\n\nThe GISAID nomenclature classifies sequences based on key mutations that define particular groups of sequences in the global phylogeny. It also complements its classification by borrowing information from the Pango nomenclature system.\nThe Pango nomenclature is based on the phylogeny of SARS-CoV-2 and defined as groups of sequences that share a common ancestor and a distinctive sequence feature (for example, all share the same single nucleotide change). This nomenclature is also sometimes referred as “Rambaut et al. 2020” after the respective publication.\nThe Nextstrain nomenclature is slightly more informal than the above, its main purpose being to facilitate public health discussions. Despite this, it is still informed by the phylogenetic placement of sequences in a tree and therefore has a large overlap with the Pango nomenclature.\n\nWe will learn more about how sequences are classified into lineages and variants of concern in the section about Lineage Assignment.\n\n\n\n\n\n\nWhat is the difference: strain, lineage, clade, variant?\n\n\n\nThese terms are sometimes used interchangeably in informal conversations about the different forms of SARS-CoV-2. For our purposes, these are the definitions we will use:\nA strain is a group of viruses that are sufficiently diverged from others, so that they are quite distinct at a sequence level as well as in their biological properties. SARS-CoV-2 is still considered to be a single strain. Examples of other coronavirus strains are SARS-CoV and MERS-CoV.\nThe terms lineage and clade are somewhat similar, in that both represent groups of similar sequences, inferred from a phylogenetic analysis and sharing a common ancestor. Their main difference (at least in the current SARS-CoV-2 nomenclature systems) is the level of resolution. Clades tend to be more broadly defined and therefore include more sequences within it. They are useful to discuss long-term trends during a pandemic. Lineages have a finer resolution, being useful to identify patterns related to a recent outbreak.\nSince a phylogenetic tree is inherently hierarchical, there isn’t always a clear-cut way of defining where one lineage/clade starts and another ends. That is why lineage classification is a partially manual process, involving human curation by experts.\nFinally, the term variant is usually used to refer to WHO’s variants of interest or variants of concern (e.g. the Alpha, Delta and Omicron variants). Variants are distinct from each other by the combination of all sequence changes in their genomes. The term “variant” can be ambiguous when used in the field of bioinformatics, and we return to this in the section about Lineage Assigment"
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#the-gisaid-database",
+ "href": "materials/01-intro/01-surveillance.html#the-gisaid-database",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.3 The GISAID Database",
+ "text": "1.3 The GISAID Database\nThe widespread use of genome sequences would have not been possible without the ability to centrally collect these sequences and make them available to researchers and public health professionals. The main repository used to deposit SARS-CoV-2 sequences is the database managed by the GISAID Initiative. This allows sharing the sequencing data as well as metadata associated with it (such as dates of collection, geo-location, patient information, etc.). For example, the outbreak.info website that we just used in the exercise above directly pulls data from GISAID to update its reports on a daily basis.\nAt the end of this course you too will be able to contribute to this database, by producing full genome sequences assembled from sequencing data.\n\n\n\n\n\n\nCreate a GISAID account\n\n\n\nGo to the GISAID registration page and create an account, so you can gain access to the data stored in GISAID as well as the ability to submit your own sequences in the future."
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#sars-cov-2-sequencing",
+ "href": "materials/01-intro/01-surveillance.html#sars-cov-2-sequencing",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.4 SARS-CoV-2 Sequencing",
+ "text": "1.4 SARS-CoV-2 Sequencing\nRoutine SARS-CoV-2 sequencing is done with a method generally referred to as amplicon sequencing. This method relies on amplifying the genetic material using polymerase chain reaction (PCR) with a panel of primers designed against the known SARS-CoV-2 genome.\nOne of the most popular methods for preparing virus RNA for sequencing is amplicon sequencing. This method consists of amplifying the genetic material by PCR using a panel of primers that covers the entire genome of the target virus. This is required, since the starting amount of viral genetic material in a sample is typically very low (most of the material will belong to the host - in this case, human RNA).\nThe most popular protocol for amplicon sequencing has been developed by the ARTIC network, whose aim is to develop standardised protocols for viral sample processing. The group has designed and tested a panel of primers that work well to amplify the SARS-CoV-2 genome in a mostly unbiased way. This is a challenging task, as the protocol involves pooling hundreds of primers together in a single reaction! Also, as the virus mutates in the population, primers that used to work in the original template genome may no longer work in the new variants circulating in the population (“primer erosion”). Therefore, the ARTIC primers have gone through several versions, which are updated and optimised to work on the most common circulating lineages. These are called “primer schemes” and are made publicly available in a public repository.\n\n\n\nSchematic of the ARTIC protocol. The RNA is reverse-transcribed to cDNA, then two pools of primers (that cover the entire SARS-CoV-2 genome) are used to PCR-amplify the material. This material is used to prepare sequencing libraries for the relevant platform that will be used downstream (Illumina or Nanopore). Source: Gohl et al. 2020\n\n\nBesides amplicon sequencing, other methods can also be used to obtain SARS-Cov-2 genomes:\n\nMetagenomic RNA sequencing was the method used to assemble the first SARS-CoV-2 genome and one of the first sequences in Cambodia. This method consists of extracting viral RNA (using commercially available kits) followed by high-throughput RNA-seq. The resulting sequences are then compared with nucleotide databases of known organisms, and viral sequences selected for de-novo assembly. This approach is suitable when the virus sequence is unknown, but requires a high sequencing depth, increasing its costs.\nSequence capture protocols are also available, whereby the samples are enriched for the target virus by using a panel of probes against the SARS-CoV-2 genome, followed by sequencing and reference-based assembly. This approach is more similar to amplicon sequencing, as it works by enriching the sample for the known virus.\n\n\n\n\nSimplified diagram of the main steps involved in metagenomic and amplicon sequencing approaches. In amplicon sequencing the cDNA material is first PCR-amplified with virus-specific primers to enrich the sample, followed by sequencing and downstream bioinformatic analysis. With the metagenomic approach the mixed cDNA material is sequenced and the resulting sequences are bioinformatically separated between known sequences (from other organisms) and unknown sequences. The unknown sequences can then be de-novo assembled into a new genome.\n\n\nDespite these alternative methods, amplicon sequencing remains one of the most popular methods for large-scale viral surveillance due to its low cost and high-throughput. The data generated from this method will be the focus of this course.\n\n\n\n\n\n\nIllumina or Nanopore?\n\n\n\nBoth of these sequencing platforms can be used to sequence amplicon samples.\nNanopore platforms allow sequencing 96 samples at a time, are readily available as portable devices, and have fast run times. This gives them great flexibility, making them an excellent solution for rapidly building sequencing capacity in a lab.\nBy comparison, Illumina platforms give higher throughput, are cheaper to run per sample and have lower error rates. However, they require substantial upfront cost to setup and equip in the lab and take longer to run.\n\n\n\n\n\n\n\n\nAlternative Amplicon Sequencing Protocols\n\n\n\nAlthough the ARTIC protocol is one of the most popular used in routine SARS-CoV-2 sequencing, there are alternative sets of primers and protocols available.\nFor example, Thermo Fisher has the Ion AmpliSeq SARS-CoV-2 kit, designed to work with Ion Torrent sequencing platforms.\nAn alternative protocol that has been developed by the community is the midnight protocol. This protocol consists of amplifying larger PCR fragments, thus requiring fewer primer pairs than the ARTIC protocol. This leads to a lower complexity in the multiplex PCR reaction and fewer chances of PCR dropout due to mis-priming againts new variants. However, because it uses longer PCR fragments, it can only be used with long-read sequencing (Nanopore) and not short-read sequencing (Illumina).\nThe Coronavirus Method Development Community on the protocols.io platform is a good source of alternative sequencing protocols for SARS-CoV-2."
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#sample-collection",
+ "href": "materials/01-intro/01-surveillance.html#sample-collection",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.5 Sample Collection",
+ "text": "1.5 Sample Collection\nThere are two big considerations when collecting samples for sequencing:\n\nIs there enough viral material in the sample (viral load)?\nDid I collect all the necessary information about each sample (metadata)?\n\n\n1.5.1 Viral Load\nWhen collecting patient samples for sequencing, it is important to quantify the viral load in the sample. This is usually done by quantitative RT-PCR (RT-qPCR), whereby the amplification of the samples is monitored by measuring a dye that is incorporated during the PCR reaction. This results in amplification curves, which reflect the amount of RNA present in the sample. The more PCR cycles are needed to saturate the signal, the lower the amount of virus in the sample.\n\n\n\nOverview of RT-qPCR quantification of viral load in a sample. Adapted from Smyrlaki et al. 2020\n\n\nThe result of RT-qPCR is usually expressed as the PCR cycle at which a particular threshold in the amplification curve is reached, the Ct value. Generally, samples with Ct > 30 are not worth sequencing, as their genome completeness is likely going to be low due to the low amounts of starting material.\n\n\n\nRelationship between RT-qPCR Ct value and resulting genome completeness. Source: Jared Simpson’s talk at CLIMB BIG DATA workshop\n\n\n\n\n1.5.2 Metadata\nOne important consideration when collecting samples, is to record as much information as possible about each sample. The Public Health Alliance for Genomic Epidemiology (PHA4GE) coalition provides several guidelines and a protocol to aid in metadata collection. There are also essential metadata needed to upload new SARS-CoV-2 genome sequences to the GISAID database (Figure).\n\n\n\nSnapshot of the GISAID upload template metadata sheet.\n\n\nTwo key pieces of information for genomic surveillance are the date of sample collection and the geographic location of that sample. This information can be used to understand which variants are circulating in an area at any given time.\nPrivacy concerns need to be considered when collecting and storing sensitive data. However, it should be noted that sensitive data can still be collected, even if it is not shared publicly (e.g. via the GISAID database). Such sensitive information may still be useful for the relevant public health authorities, who may use those sensitive information for a finer analysis of the data. For example, epidemiological analysis will require individual-level metadata (“person, place, time”) to be available, in order to track the dynamics of transmission within a community.\nThis is the general advice when it comes to metadata collection: record as much information about each sample as possible!"
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#sars-cov-2-bioinformatics",
+ "href": "materials/01-intro/01-surveillance.html#sars-cov-2-bioinformatics",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.6 SARS-CoV-2 Bioinformatics",
+ "text": "1.6 SARS-CoV-2 Bioinformatics\nWhat bioinformatic skills do we need in order to analyse SARS-CoV-2 genome sequencing data? While there are several software tools that have been specifically developed for SARS-CoV-2 analysis (and we will see some of them in this course), there is a set of foundational skills that are applicable to any bioinformatics application:\n\nThe use of the Unix command line. Linux is the most common operating system for computational work and most of the bioinformatic software only runs on it.\nGetting familiar with common file formats in bioinformatics. This includes files to store nucleotide sequences, sequence alignments to a reference genome, gene annotations, phylogenetic trees, amongst others.\nUnderstand software tools’ documentation and how to configure different options when running our analyses.\n\nWe will turn to these topics in the following sessions."
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#exercises",
+ "href": "materials/01-intro/01-surveillance.html#exercises",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.7 Exercises",
+ "text": "1.7 Exercises\n\n\n\n\n\n\nWHO Variants\n\n\n\n\n\n\n\n\nLooking through the WHO variants page, can you find what the difference is between a Variant of Interest (VOI) and a Variant of Concern (VOC)?\nCan you find the correspondence between the VOCs and their respective lineages in other classification systems (GISAID, Nextstrain and Pango)?\nGo to the outbreak.info website and search for a country of your choice (for example, your country of origin or where you live).\nHow many sequences are available from the last 360 days? (Note: by default only the last 60 days are shown. You can change this in the text box found on the right of the report page.)\nWhat were the most common lineages of the virus in circulation in the last 360 days? Do you notice sharp changes in the frequency of WHO Variants of Concern?\n\n\n\n\n\n\n\n\n\n\n\n\nMetadata\n\n\n\n\n\n\n\nOpen the folder under “Course_Materials > 01-Unix > metadata”, where you will find several CSV files with information about samples that were sequenced and will be analysed by us later in the course. Double-click to open one of these files (any of them is fine), which should open them on a spreadsheet software (on our training machines it is LibreOffice, but Excel would also work).\nThe column names of these files are based on the PHA4GE nomenclature system.\n\nGo to the PHA4GE metadata collection protocol and read throught the first steps to find the link to the collection templates.\nFrom that link, go to the Supporting Materials to find the “field mappings” spreadsheet, which matches naming conventions between PHA4GE and other databases such as GISAID.\nBased on this information, can you match the column names from our sample information table to the fields required by GISAID?\nWhat do you think is the naming convention for the isolate name? How would you adjust this when uploading the data to GISAID?\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nThe first step of this protocol provides with a link to a GitHub repository containing several template files: https://github.com/pha4ge/SARS-CoV-2-Contextual-Data-Specification.\nIf we follow that link, we will see several spreadsheet files (for Excel or LibreOffice) with templates that can be used to record samples metadata. In the “Supporting Materials” we find a link to “PHA4GE to WHO and sequence repository field mappings”. If we download and open that file we will see a spreadhseet with 4 tabs. Each of the tabs shows the equivalence between the PHA4GE column names and the equivalent names used by other platforms.\nLooking at the equivalence between the column names in our metadata sheets and the GISAID nomenclature, we can identify only some of them:\n\n\n\n\n\n\n\n\nOur Name\nGISAID equivalent\nComments\n\n\n\n\nsample\n\n\n\n\nsample_collection_date\nCollection date\n\n\n\ngeo_loc_country\nLocation\n\n\n\ngeo_loc_region\nLocation\nIn GISAID the field “Location” is used for “Continent / Country or Territory / Region”\n\n\nlatitude\n\n\n\n\nlongitude\n\n\n\n\nsequencing_instrument\nSequencing technology\n\n\n\nsample_collection_year\n\n\n\n\nsample_collection_month\n\n\n\n\nsample_collection_day\n\n\n\n\norganism\n\n\n\n\nisolate\nVirus name\n\n\n\nsequencing_protocol_name\n\n\n\n\namplicon_pcr_primer_scheme\n\n\n\n\nsequencing_protocol\n\n\n\n\n\nLooking at this spreadhseet, we can also see this explanation about the “Virus Name” field:\n\nWhile the meanings and structures of the GISAID field “Virus name” and the PHA4GE field “isolate” overlap, GISAID requires a slightly different structure for Virus name. e.g. PHA4GE structure: SARS-CoV-2/country/sampleID/2020, GISAID structure: hCov-19/country/sampleID/2020. Change “SARS-CoV-2” to “hCov-19” in the isolate name.\n\nSo, in order to upload our data to GISAID we would need to change the name of the virus accordingly."
+ },
+ {
+ "objectID": "materials/01-intro/01-surveillance.html#summary",
+ "href": "materials/01-intro/01-surveillance.html#summary",
+ "title": "1 SARS-CoV-2 genomic surveillance",
+ "section": "1.8 Summary",
+ "text": "1.8 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nThe sequencing of SARS-CoV-2 genomes has allowed the tracking of new variants throughout the pandemic.\nThe World Health Organisation defines Variants of Concern as SARS-CoV-2 forms with characteristics of public health concern. This includes increased transmissibility, virulence, disease symptoms or vaccine resistance.\nGISAID, Pango and Nextstrain are organisations whose work includes the classification of SARS-CoV-2 genomes into groups that may later be classified as variants of concern by WHO.\nGISAID also plays a key role as the main database for assembled SARS-CoV-2 genomes, submitted by the community.\nRoutine SARS-CoV-2 sequencing is usually done by amplicon sequencing (amplifying an infected sample by PCR using primers that cover the entire genome). The most popular protocol has been developed by the ARTIC network.\nOther methods of sequencing include metagenomic and sequence capture. However, these methods require more sequencing and therefore are not commonly used for sequencing at a population scale.\nBoth Illumina and Nanopore platforms can be used for sequencing SARS-CoV-2 amplicon samples.\nMetadata collection is essential for interpreting the results of the sequencing.\n\nFor genomic surveillance purposes recording of geographic location and date of sampling are crucial.\nOther useful information includes details about the sequencing (e.g. sample preparation protocols and sequencing platforms used)."
+ },
+ {
+ "objectID": "materials/01-intro/02-ngs.html#next-generation-sequencing",
+ "href": "materials/01-intro/02-ngs.html#next-generation-sequencing",
+ "title": "2 Introduction to NGS",
+ "section": "2.1 Next Generation Sequencing",
+ "text": "2.1 Next Generation Sequencing\nThe sequencing of genomes has become more routine due to the rapid drop in DNA sequencing costs seen since the development of Next Generation Sequencing (NGS) technologies in 2007. One main feature of these technologies is that they are high-throughput, allowing one to more fully characterise the genetic material in a sample of interest.\nThere are three main technologies in use nowadays, often referred to as 2nd and 3rd generation sequencing:\n\nIllumina’s sequencing by synthesis (2nd generation)\nOxford Nanopore, shortened ONT (3rd generation)\nPacific Biosciences, shortened PacBio (3rd generation)\n\nThe video below from the iBiology team gives a great overview of these technologies.\n\n\n\n\n\n2.1.1 Illumina Sequencing\nIllumina’s technology has become a widely popular method, with many applications to study transcriptomes (RNA-seq), epigenomes (ATAC-seq, BS-seq), DNA-protein interactions (ChIP-seq), chromatin conformation (Hi-C/3C-Seq), population and quantitative genetics (variant detection, GWAS), de-novo genome assembly, amongst many others.\nAn overview of the sequencing procedure is shown in the animation video below. Generally, samples are processed to generate so-called sequencing libraries, where the genetic material (DNA or RNA) is processed to generate fragments of DNA with attached oligo adapters necessary for the sequencing procedure (if the starting material is RNA, it can be converted to DNA by a step of reverse transcription). Each of these DNA molecule is then sequenced from both ends, generating pairs of sequences from each molecule, i.e. paired-end sequencing (single-end sequencing, where the molecule is only sequenced from one end is also possible, although much less common nowadays).\nThis technology is a type of short-read sequencing, because we only obtain short sequences from the original DNA molecules. Typical protocols will generate 2x50bp to 2x250bp sequences (the 2x denotes that we sequence from each end of the molecule).\n\n\n\n\nThe main advantage of Illumina sequencing is that it produces very high-quality sequence reads (current protocols generate reads with an error rate of less than <1%) at a low cost. However, the fact that we only get relatively short sequences means that there are limitations when it comes to resolving particular problems such as long sequence repeats (e.g. around centromeres or transposon-rich areas of the genome), distinguishing gene isoforms (in RNA-seq), or resolving haplotypes (combinations of variants in each copy of an individual’s diploid genome).\n\n\n2.1.2 Nanopore Sequencing\nNanopore sequencing is a type of long-read sequencing technology. The main advantage of this technology is that it can sequence very long DNA molecules (up to megabase-sized), thus overcoming the main shortcoming of short-read sequencing mentioned above. Another big advantage of this technology is its portability, with some of its devices designed to work via USB plugged to a standard laptop. This makes it an ideal technology to use in situations where it is not possible to equip a dedicated sequencing facility/laboratory (for example, when doing field work).\n\n\n\nOverview of Nanopore sequencing showing the highly-portable MinION device. The device contains thousands of nanopores embeded in a membrane where current is applied. As individual DNA molecules pass through these nanopores they cause changes in this current, which is detected by sensors and read by a dedicated computer program. Each DNA base causes different changes in the current, allowing the software to convert this signal into base calls.\n\n\nOne of the bigger challenges in effectively using this technology is to produce sequencing libraries that contain high molecular weight, intact, DNA. Another disadvantage is that, compared to Illumina sequencing, the error rates at higher, at around 5%.\n\n\n\n\n\n\nIllumina or Nanopore for SARS-CoV-2 sequencing?\n\n\n\nBoth of these platforms have been widely popular for SARS-CoV-2 sequencing. They can both generate data with high-enough quality for the assembly and analysis of SARS-CoV-2 genomes. Mostly, which one you use will depend on what sequencing facilities you have access to.\nWhile Illumina provides the cheapest option per sample of the two, it has a higher setup cost, requiring access to the expensive sequencing machines. On the other hand, Nanopore is a very flexible platform, especially its portable MinION devices. They require less up-front cost allowing getting started with sequencing very quickly in a standard molecular biology lab."
+ },
+ {
+ "objectID": "materials/01-intro/02-ngs.html#sequencing-analysis",
+ "href": "materials/01-intro/02-ngs.html#sequencing-analysis",
+ "title": "2 Introduction to NGS",
+ "section": "2.2 Sequencing Analysis",
+ "text": "2.2 Sequencing Analysis\nIn this section we will demonstrate two common tasks in sequencing data analysis: sequence quality control and mapping to a reference genome. There are many other tasks involved in analysing sequencing data, but looking at these two examples will demonstrate the principles of running bioinformatic programs. We will later see how bioinformaticians can automate more complex analyses in the consensus assembly section.\nOne of the main features in bioinformatic analysis is the use of standard file formats. It allows software developers to create tools that work well with each other. For example, the raw data from Illumina and Nanopore platforms is very different: Illumina generates images; Nanopore generates electrical current signal. However, both platforms come with software that converts those raw data to a standard text-based format called FASTQ.\n\n2.2.1 FASTQ Files\nFASTQ files are used to store nucleotide sequences along with a quality score for each nucleotide of the sequence. These files are the typical format obtained from NGS sequencing platforms such as Illumina and Nanopore (after basecalling).\nThe file format is as follows:\n@SEQ_ID <-- SEQUENCE NAME\nAGCGTGTACTGTGCATGTCGATG <-- SEQUENCE\n+ <-- SEPARATOR\n%%).1***-+*''))**55CCFF <-- QUALITY SCORES\nIn FASTQ files each sequence is always represented across 4 lines. The quality scores are encoded in a compact form, using a single character. They represent a score that can vary between 0 and 40 (see Illumina’s Quality Score Encoding). The reason single characters are used to encode the quality scores is that it saves space when storing these large files. Software that work on FASTQ files automatically convert these characters into their score, so we don’t have to worry about doing this conversion ourselves.\nThe quality value in common use is called a Phred score and it represents the probability that the respective base is an error. For example, a base with quality 20 has a probability \\(10^{-2} = 0.01 = 1\\%\\) of being an error. A base with quality 30 has \\(10^{-3} = 0.001 = 0.1\\%\\) chance of being an error. Typically, a Phred score threshold of >20 or >30 is used when applying quality filters to sequencing reads.\nBecause FASTQ files tend to be quite large, they are often compressed to save space. The most common compression format is called gzip and uses the extension .gz. To look at a gzip file, we can use the command zcat, which decompresses the file and prints the output as text.\nFor example, we can use the following command to count the number of lines in a compressed FASTQ file:\nzcat sequences.fq.gz | wc -l\nIf we want to know how many sequences there are in the file, we can divide the result by 4 (since each sequence is always represented across four lines).\n\n\n2.2.2 FASTQ Quality Control\nOne of the most basic tasks in Illumina sequence analysis is to run a quality control step on the FASTQ files we obtained from the sequencing machine.\nThe program used to assess FASTQ quality is called FastQC. It produces several statistics and graphs for each file in a nice report that can be used to identify any quality issues with our sequences.\nThe basic command to run FastQC is:\nfastqc --outdir PATH_TO_OUTPUT_DIRECTORY PATH_TO_SEQUENCES\nFastQC can process several samples at once, and often we can use the * wildcard to do this. We will see an example of this in the following exercise.\nEach FastQC HTML report contains a section with a different quality assessment plot. Each of these are explained in the online documentation:\n\nBasic statistics\nPer base sequence quality\nPer sequence quality scores\nPer base sequence content\nPer sequence GC content\nPer base N content\nSequence length distribution\nSequence duplication levels\nOverrepresented sequences\nAdapter content\nPer tile sequence quality\n\nFor example, looking at the “Per base sequence quality” section for one of our samples, we can see a very high quality score, which is typical of Illumina data nowadays.\n\n\n\nSequence quality plot from FastQC for one of our samples. The blue line shows the average across all samples. This sample is very high quality as all sequences have quality > 20 across the entire length of the reads.\n\n\n\n\n\n\n\n\nQuality Control Nanopore Reads\n\n\n\nAlthough FastQC can run its analysis on any FASTQ files, it has mostly been designed for Illumina data. You can still run FastQC on basecalled Nanopore data, but some of the output modules may not be as informative. FastQC can also run on FAST5 files, using the option --nano.\nYou can also use MinIONQC, which takes as input the sequence_summary.txt file, which is a standard output file from the Guppy software used to convert Nanopore electrical signal to sequence calls.\n\n\n\n\n2.2.3 Read Mapping\nA common task in processing sequencing reads is to align them to a reference genome, which is typically referred to as read mapping or read alignment. We will continue exemplifying how this works for Illumina data, however the principle is similar for Nanopore data (although the software used is often different, due to the higher error rates and longer reads typical of these platforms).\nGenerally, these are the steps involved in read mapping (figure below):\n\nGenome Indexing | Because reference genomes can be quite long, most mapping algorithms require that the genome is pre-processed, which is called genome indexing. You can think of a genome index in a similar way to an index at the end of a textbook, which tells you in which pages of the book you can find certain keywords. Similarly, a genome index is used by mapping algorithms to quickly search through its sequence and find a good match with the reads it is trying to align against it. Each mapping software requires its own index, but we only have to generate the genome index once.\nRead mapping | This is the actual step of aligning the reads to a reference genome. There are different popular read mapping programs such as bowtie2 or bwa. The input to these programs includes the genome index (from the previous step) and the FASTQ file(s) with reads. The output is an alignment in a file format called SAM (text-based format - takes a lot of space) or BAM (compressed binary format - much smaller file size).\nBAM Sorting | The mapping programs output the sequencing reads in a random order (the order in which they were processed). But, for downstream analysis, it is good to sort the reads by their position in the genome, which makes it faster to process the file.\nBAM Indexing | This is similar to the genome indexing we mentioned above, but this time creating an index for the alignment file. This index is often required for downstream analysis and for visualising the alignment with programs such as IGV.\n\n\n\n\nDiagram illustrating the steps involved in mapping sequencing reads to a reference genome. Mapping programs allow some differences between the reads and the reference genome (red mutation shown as an example). Before doing the mapping, reads are usually filtered for high-quality and to remove any sequencing adapters. The reference genome is also indexed before running the mapping step. The mapped file (BAM format) can be used in many downstream analyses. See text for more details.\n\n\nWe have already prepared the SARS-CoV-2 genome index for the bowtie2 aligner. We have also prepared a shell script with the code to run the three steps above as an example. Let’s look at the content of this file (you can open it with nano scripts/mapping.sh):\n# mapping\nbowtie2 -x resources/reference/bowtie2/sarscov2 -1 data/reads/ERR6129126_1.fastq.gz -2 data/reads/ERR6129126_2.fastq.gz --threads 5 | samtools sort -o results/bowtie2/ERR6129126.bam -\n\n# index mapped file\nsamtools index results/bowtie2/ERR6129126.bam\n\n# obtain some mapping statistics\nsamtools stats results/bowtie2/ERR6129126.bam > results/bowtie2/ERR6129126.stats.txt\nIn the first step, mapping, we are using two tools: bowtie2 and samtools. bowtie2 is the mapping program and samtools is a program used to manipulate SAM/BAM alignment files. In this case we used the | pipe to send the output of bowtie2 directly to samtools:\n\n-x is the prefix of the reference genome index.\n-1 is the path to the first read in paired-end sequencing.\n-2 is the path to the second read in paired-end sequencing.\n--threads 5 indicates we want to use 5 CPUs (or threads) to do parallel processing of the data.\n| is the pipe that sends the output from bowtie2 to samtools sort.\n-o is the name of the output file. By setting the file extension of this file to .bam, samtools will automatically save the file in the compressed format (which saves a lot of space).\nThe - symbol at the end of the samtools command indicates that the input is coming from the | pipe.\n\nWe also have a step that creates an index file for the BAM file using samtools index. This creates a file with the same name and .bai extension.\nFinally, the script also contains a step that collects some basic statistics from the alignment, which we save in a text file. We will see how this file can be used to produce a quality control report below.\n\n\n2.2.4 Visualising BAM Files in IGV\nOne thing that can be useful is to visualise the alignments produced in this way. We can use the program IGV (Integrative Genome Viewer) to do this:\n\nOpen IGV and go to File → Load from file….\nIn the file browser that opens go to the folder results/bowtie2/ and select the file ERR6129126.bam to open it.\nGo back to File → Load from file… and this time load the BED files containing the primer locations. These can be found in resources/primers/artic_primers_pool1.bed and resources/primers/artic_primers_pool2.bed.\n\nThere are several ways to search and browse through our alignments, exemplified in the figure below.\n\n\n\nScreenshot IGV program. The search box at the top can be used to go to a specific region in the format “CHROM:START-END”. In the case of SARS-CoV-2 there is only one “chromosome” called NC_045512.2 (this is the name of the reference genome), so if we wanted to visualise the region between positions 21563 and 25384 (the Spike gene) we would write “NC_045512.2:21563-25384”.\n\n\n\n\n2.2.5 Quality Reports\nWe’ve seen the example of using the program FastQC to assess the quality of our FASTQ sequencing files. And we have also seen an example of using the program samtools stats to obtain some quality statistics of our read mapping step.\nWhen processing multiple samples at once, it can become hard to check all of these quality metrics individually for each sample. This is the problem that the software MultiQC tries to solve. This software automatically scans a directory and looks for files it recognises as containing quality statistics. It then compiles all those statistics in a single report, so that we can more easily look across dozens or even hundreds of samples at once.\nHere is the command to run MultiQC and compile several quality statistics into a single report:\nmkdir results/multiqc\nmultiqc --outdir results/multiqc/ results/\nMultiQC generates a report, in this example in results/multiqc/multiqc_report.html. From this report we can get an overview of the quality across all our samples.\n\n\n\nSnapshot of some of the report sections from MultiQC. In this example we can see the “General Statistics” table and the “Sequence Quality Histograms” plot. One of the samples has lower quality towards the end of the read compared to other samples (red line in the bottom panel).\n\n\nFor example, from the section “General Statistics” we can see that the number of reads varies a lot between samples. Sample ERR5926784 has around 0.1 million reads, which is substantially lower than other samples that have over 1 million reads. This may affect the quality of the consensus assembly that we will do afterwards.\nFrom the section “Sequence Quality Histograms”, we can see that one sample in particular - ERR5926784 - has lower quality in the second pair of the read. We can open the original FastQC report and confirm that several sequences even drop below a quality score of 20 (1% change of error). A drop in sequencing quality towards the end of a read can often happen, especially for longer reads. Usually, analysis workflows include a step to remove reads with low quality so these should not affect downstream analysis too badly. However, it’s always good to make a note of potentially problematic samples, and see if they produce lower quality results downstream."
+ },
+ {
+ "objectID": "materials/01-intro/02-ngs.html#bioinformatic-file-formats",
+ "href": "materials/01-intro/02-ngs.html#bioinformatic-file-formats",
+ "title": "2 Introduction to NGS",
+ "section": "2.3 Bioinformatic File Formats",
+ "text": "2.3 Bioinformatic File Formats\nLike we said above, bioinformatics uses many standard file formats to store different types of data. We have just seen two of these file formats: FASTQ for sequencing reads and BAM files to store reads mapped to a genome.\nAnother very common file format is the FASTA file, which is the format that our reference genome is stored as. The consensus sequences that we will generate are also stored as FASTA files. We detail this format below, but there are many other formats. Check out our appendix page on File Formats to learn more about them.\n\n2.3.1 FASTA Files\nAnother very common file that we should consider is the FASTA format. FASTA files are used to store nucleotide or amino acid sequences.\nThe general structure of a FASTA file is illustrated below:\n>sample01 <-- NAME OF THE SEQUENCE\nAGCGTGTACTGTGCATGTCGATG <-- SEQUENCE ITSELF\nEach sequence is represented by a name, which always starts with the character >, followed by the actual sequence.\nA FASTA file can contain several sequences, for example:\n>sample01\nAGCGTGTACTGTGCATGTCGATG\n>sample02\nAGCGTGTACTGTGCATGTCGATG\nEach sequence can sometimes span multiple lines, and separate sequences can always be identified by the > character. For example, this contains the same sequences as above:\n>sample01 <-- FIRST SEQUENCE STARTS HERE\nAGCGTGTACTGT\nGCATGTCGATG\n>sample02 <-- SECOND SEQUENCE STARTS HERE\nAGCGTGTACTGT\nGCATGTCGATG\nTo count how many sequences there are in a FASTA file, we can use the following command:\ngrep \">\" sequences.fa | wc -l\nIn two steps:\n\nfind the lines containing the character “>”, and then\ncount the number of lines of the result.\n\nWe will see FASTA files several times throughout this course, so it’s important to be familiar with them."
+ },
+ {
+ "objectID": "materials/01-intro/02-ngs.html#exercises",
+ "href": "materials/01-intro/02-ngs.html#exercises",
+ "title": "2 Introduction to NGS",
+ "section": "2.4 Exercises",
+ "text": "2.4 Exercises\n\n\n\n\n\n\nSequence quality control: FASTQC\n\n\n\n\n\n\n\nIn the course materials directory 02-ngs/ we have several FASTQ files that we will use to assemble SARS-CoV-2 genomes. But first, we will run FastQC to check the quality of these files.\nThis is the basic command we could use in our samples:\nfastqc --outdir results/fastqc data/reads/*.fastq.gz\n\nCreate the output directory for the analysis (results/fastqc).\n\n\nHint\n\nThe command to create directories is mkdir. By default, the mkdir directory only creates one directory at a time. In this case we need to create first the results directory and then the results/fastqc within it. Alternatively, both directories can be created at once using the -p option.\n\nModify the fastqc command shown above to add an option to run the analysis using 8 threads in parallel (or CPUs). Check the tool’s help (fastqc --help) to see what the option to do this is called.\nRun the command. You will know it is running successfully because it prints progress of its analysis on the screen.\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nFirst, we can create a directory to output our results:\nmkdir -p results/fastqc\nThe -p option ensures that both directories are created in one step. Otherwise, since the parent directory results did not exist, mkdir would throw an error.\nTo check the options available with FastQC we can run fastqc --help to get the complete documentation. As we scroll through the options, we can see the relevant one for running the analysis in parallel:\n-t --threads Specifies the number of files which can be processed\n simultaneously. Each thread will be allocated 250MB of\n memory so you shouldn't run more threads than your\n available memory will cope with, and not more than\n 6 threads on a 32 bit machine\nAlthough the documentation is a little technical, this means that if we have multiple CPUs available on our computer, we can set this option to allow multiple files to be processed in parallel. Our training machines have 8 CPUs, so we can run the command as follows:\nfastqc -t 8 --outdir results/fastqc data/reads/*.fastq.gz\nThe analysis report generated by FastQC is given as a .html file (opens in a web browser). We will go through the details of this below."
+ },
+ {
+ "objectID": "materials/01-intro/02-ngs.html#summary",
+ "href": "materials/01-intro/02-ngs.html#summary",
+ "title": "2 Introduction to NGS",
+ "section": "2.5 Summary",
+ "text": "2.5 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nIllumina sequencing produces short reads (50bp - 200bp), typically from both ends of a DNA fragment. It is a comparatively cheap sequencing platform which produces very high-quality sequences.\nNanopore sequencing produces very long reads (typically hundreds of kilobases long). It is comparatively more expensive and has higher error rates. However, it is more flexible with some of its platforms being fully portable.\nSequencing reads are stored in a file format called FASTQ. This file contains both the nucleotide sequence and quality of each base.\nThe quality of Illumina sequence reads can be assessed using the software FastQC.\nOne common task in bioinformatics is to align or map reads to a reference genome. This involves:\n\nCreating a genome index - this only needs to be done once.\nMapping the reads to the reference genome (e.g. using bowtie2) - the output is in SAM format.\nSorting the reads in the mapped file (using samtools sort) - the output is in BAM format.\nIndexing the BAM alignment file (using samtools index).\n\nThe software MultiQC can be used to generate a single reports that compiles statistics across several samples.\nBioinformatics uses many standard file formats. One of the most common ones is the FASTA format, which is used to store nucleotide or amino acid sequences (no quality information is contained in these files). This is a standard format that assembled genomes are stored as."
+ },
+ {
+ "objectID": "materials/01-intro/03-workflows.html",
+ "href": "materials/01-intro/03-workflows.html",
+ "title": "3 Bioinformatic workflows",
+ "section": "",
+ "text": "Warning\n\n\n\nUnder development"
+ },
+ {
+ "objectID": "materials/02-isolates/01-consensus.html#sec-consensus",
+ "href": "materials/02-isolates/01-consensus.html#sec-consensus",
+ "title": "4 Consensus assembly",
+ "section": "4.1 SARS-CoV-2 Consensus Assembly",
+ "text": "4.1 SARS-CoV-2 Consensus Assembly\nAs we discussed earlier in the course, the starting material for sequencing SARS-CoV-2 samples from infected patients is PCR-amplified DNA generated with a panel of primers that covers the whole SARS-CoV-2 genome (for example the primers developed by the ARTIC network). This material can then be sequenced using either Illumina or Nanopore platforms.\nAlthough different sotware tools are used depending on which kind of sequencing platform was used, the main goal is the same: to align the sequencing reads to the reference genome, and identify any DNA changes (SNPs or Indels) relative to the reference genome (Wuhan-Hu-1). This is called consensus assembly, since we are assembling the genome of our sample from the PCR-amplified fragments and generating a consensus sequence based on changes present in several reads covering a particular position of the genome.\nThe general data processing steps are:\n\nFilter high-quality sequencing reads.\nMap the reads to the Wuhan-Hu-1 reference genome.\nTrim the primers from the aligned reads based on the primer location file (BED file).\nPerform variant calling (SNPs and indels) to identify changes relative to the reference sequence.\nGenerate a consensus sequence for the sample based on those variants.\n\n\n\n\nOverview of the consensus assembly procedure from amplicon sequencing reads. In this schematic, each read spans the whole length of a PCR amplicon, which is what is expected from Nanopore reads. With Illumina data, there would be two pairs of reads starting at each end of the PCR amplicon.\n\n\n\n\n\n\n\n\nPrimer trimming\n\n\n\nPrimer trimming is a key step of the data processing, otherwise SNPs might be missed at the primer sites, on the final consensus sequence. This is because the primer sequence is retained during PCR instead of the original sequence of the sample. Because the PCR amplicons overlap with each other, we can trim the primers from each read and do variant calling after trimming. An example of this is shown in the Figure above."
+ },
+ {
+ "objectID": "materials/02-isolates/01-consensus.html#bioinformatic-workflowspipelines",
+ "href": "materials/02-isolates/01-consensus.html#bioinformatic-workflowspipelines",
+ "title": "4 Consensus assembly",
+ "section": "4.2 Bioinformatic Workflows/Pipelines",
+ "text": "4.2 Bioinformatic Workflows/Pipelines\nAs can already be seen from the brief description above, bioinformatic analyses always involve multiple steps where data is gathered, cleaned and integrated to give a final set of processed files of interest to the user. These sequences of steps are called a workflow or pipeline. As analyses become more complex, pipelines may include the use of many different software tools, each requiring a specific set of inputs and options to be defined. Furthermore, as we want to chain multiple tools together, the inputs of one tool may be the output of another, which can become challenging to manage.\nAlthough it is possible to code such workflows using shell scripts, these often don’t scale well across different users and compute setups. To overcome these limitations, dedicated workflow/pipeline management software packages have been developed to help standardise pipelines and make it easier for the user to process their data.\n\nTwo of the most popular workflow software packages are Snakemake and Nextflow. We will not cover how to develop workflows with these packages, but rather how to use an existing workflow to generate consensus sequences from SARS-CoV-2 data.\n\nWhy Use a Standardised Workflow?\nThese are some of the key advantages of using a standardised workflow for our analysis:\n\nFewer errors - because the workflow automates the process of managing input/output files, there are less chances for errors or bugs in the code to occur.\nConsistency and reproducibility - analysis ran by different people should result in the same output, regardless of their computational setup.\nSoftware installation - all software dependencies are automatically installed for the user using solutions such as Conda, Docker and Singularity (more about these in a later section of the course).\nScalability - workflows can run on a local desktop or scale up to run on high performance compute clusters.\nCheckpoint and resume - if a workflow fails in one of the tasks, it can be resumed at a later time."
+ },
+ {
+ "objectID": "materials/02-isolates/01-consensus.html#sec-viralrecon",
+ "href": "materials/02-isolates/01-consensus.html#sec-viralrecon",
+ "title": "4 Consensus assembly",
+ "section": "4.3 SARS-CoV-2 Pipeline",
+ "text": "4.3 SARS-CoV-2 Pipeline\nTo generate consensus SARS-CoV-2 genomes from these data, we will use a pipeline that was developed by the Nextflow core team called nf-core/viralrecon (which was itself inspired by a previous pipeline from the Connor Lab). Its objective is to harmonise the assembly of SARS-CoV-2 genomes from both Illumina and Nanopore amplicon sequencing data. It can also work with metagenomic data, which we will not cover in this workshop. This pipeline therefore includes different sub-pipelines, which are launched depending on the type of sequence data we have. Watch the video below to learn more about the development of this pipeline.\n\n\n\n\nGenerally speaking, Nextflow pipelines are run with the command nextflow run PIPELINE_NAME, where “PIPELINE_NAME” is the name of the pipeline. Pipelines are usually shared in a public repository such as GitHub, and nextflow will automatically download the pipeline if it hasn’t been downloaded already to your computer.\nLet’s test our pipeline by looking at its help documentation:\nnextflow run nf-core/viralrecon -r 2.6.0 --help\nThe command should print a long list of options available with this pipeline. For pipelines developed by the Nextflow core team you can also consult the documentation available online, which is easier to read: nf-co.re/viralrecon. This page includes many details about the pipeline: which tools are used in different steps of the data processing, how to use the pipeline for different types of data, a detailed documentation of all the options of the pipeline and explanation of the output files generated by it.\nBelow, we give an overview of the pipelines used for Illumina and Nanopore amplicon data.\n\n\n\n\n\n\nReference Genome and Primer Locations\n\n\n\nThe Wuhan-Hu-1 reference genome sequence and the amplicon primer locations (in BED file format) can all be found on the ARTIC Primer Schemes repository. The pipeline we are using takes care of downloading these files for us automatically, however it can be useful to know where to find them, in case you want to use other tools that require these files.\n\n\n\nIlluminaNanopore (FASTQ)Nanopore (FAST5)\n\n\nThe Illumina sub-workflow is based on several standard bioinformatic tools and, importantly, on the iVar software, which was developed for analysing amplicon-based sequencing data.\n\n\n\nSchematic of the key steps in the Illumina sub-workflow.\n\n\nTo run the pipeline on Illumina data, we use the following general command:\nnextflow run nf-core/viralrecon \n -r 2.6.0 -profile singularity \\\n --platform illumina \\\n --input SAMPLESHEET_CSV \\\n --outdir OUTPUT_DIRECTORY \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version PRIMER_VERSION \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade \nOne of the key options is --platform illumina, which makes sure that the correct sub-workflow will be used.\n\n\nClick to see more details about this sub-workflow\n\nIn summary, the steps performed by the Illumina sub-workflow are:\n\nAdapter trimming - this consists of trimming (or “cutting”) the sequences to remove low-quality bases and any Illumina adapter sequences that are present in the sequences.\nRemoving human (host) reads - when doing the sequencing it is possible that many reads are still from human material and this step removes them from the rest of the analysis.\nRead mapping - aligning (or mapping) the reads to the Wuhan-Hu-1 reference genome.\n\nThe software used for mapping is bowtie2.\nThe software samtools is used to convert the mapped file to BAM (instead of SAM) and sort the reads by coordinate (which is necessary for downstream steps).\n\nTrim Primers - primers are removed from the aligned reads using ivar trim (using the primer BED file).\nCall variants - identify SNPs and indels using ivar variants.\nAnnotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.\nMake consensus - apply the SNPs and indels from the previous step to the reference FASTA file.\n\nThere are two tools that can be used in this step: bcftools consensus (default) or ivar consensus (can be set with the option --consensus_caller ivar).\n\nLineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.\nQuality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.\n\n\n\n\nThe nanopore sub-workflow is based on the ARTIC bioinformatics protocol and uses several of the tools from the accompanying artic software package.\nThis sub-workflow is similar to the other nanopore sub-workflow, the main difference is the software used for generating a consensus sequence (medaka instead of nanopolish).\n\n\n\nSchematic of the key steps in the Medaka sub-workflow.\n\n\nTo run our pipeline on basecalled data (FASTQ files), we use the following command:\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --platform nanopore \\\n --input SAMPLESHEET_CSV \\\n --fastq_dir fastq_pass/ \\\n --outdir OUTPUT_DIRECTORY \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version PRIMER_VERSION \\\n --artic_minion_caller medaka \\\n --artic_minion_medaka_model MEDAKA_MODEL \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade \nSome of the key options are:\n\n--platform nanopore makes sure that the correct sub-workflow will be used.\n--artic_minion_caller medaka indicates we want to use the medaka program to do the variant/consensus calling (directly from the basecalled FASTQ files, rather than from the raw signal in the FAST5 files).\n--artic_minion_medaka_model specifies the model used by the guppy_basecaller software to do the basecalling. The model name follows the structure {pore}_{device}_{caller variant}_{caller version}. See more details about this in the medaka models documentation. Note: for recent versions of Guppy (>6) there is no exact matching model from medaka. The recommendation is to use the model for the latest version available; a list of supported models can be found on the medaka GitHub repository.\n--fastq_dir specifies the directory containing the FASTQ files. This directory should contain sub-directories for each barcoded sample following the naming convention barcodeXXXX (where X is a number between 0 and 9). By default, the guppy_basecaller software from Nanopore generates a folder called “fastq_pass” which follows this convention.\n\n\n\nClick to see more details about this sub-workflow\n\nIn summary, the steps performed by the Medaka sub-workflow are:\n\nAggregate reads from each sequencing barcode (when multiple files are availble for each barcode)\nRun the artic minion tool, which internally does several steps:\n\nMap reads to the reference genome using minimap2 (can be changed to use bwa mem with the option --artic_minion_aligner bwa).\nTrim primers from the aligned reads based on the known primer positions in the BED file (using a custom python script called align_trim.py).\nCall consensus sequences and SNP/indel variants using medaka consensus and medaka variant:\n\nPositions with less than 20x depth are treated as missing data and converted to the ambiguous base ‘N’. It is not advised to go below this threshold as the models used to call variants do not perform as well.\n\n\nAnnotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.\nLineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.\nQuality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.\n\n\n\n\nThe nanopore sub-workflow is based on the ARTIC bioinformatics protocol and uses several of the tools from the accompanying artic software package.\nThis sub-workflow is similar to the other nanopore sub-workflow, the main difference is the software used for generating a consensus sequence (nanopolish instead of medaka).\n\n\n\nSchematic of the key steps in the Nanopolish sub-workflow. (Under development)\n\n\n\n\nClick to see more details about this sub-workflow\n\nIn summary, the steps performed by the Nanopolish sub-workflow are:\n\nFilter reads to ensure they pass minimum read length thresholds:\n\nminimum length 400bp (can be changed with --min_length option)\nmaximum length 700bp (can be changed with --max_length option)\n\nRun the artic minion tool, which internally does:\n\nRead alignment to reference genome using minimap2 (can be changed to use bwa mem with the --bwa option).\nTrim primers from the aligned reads (based on the known primer positions in the BED file).\nCall consensus sequences and variants using nanopolish variants if using signal-level FAST5 files.\n\nPositions with less than 20x depth are assigned the ambiguous base ‘N’. It is not advised to go below this threshold as the models used to call variants do not perform as well.\n\n\nAnnotate variants - the called variants are annotated according to their potential effect on the genes/proteins they are located in. For example, if a mutation introduces a new stop codon, or causes a frameshift.\nLineage assignment - the consensus sequences are assigned to lineages or clades using the pangolin and nextclade programs. These are two of the main lineage/clade nomenclature systems in use. They also identify variants of concern from the consensus sequences.\nQuality control - at several steps in the pipeline different tools are used to collect quality metrics and these are compiled into a report using multiqc.\n\n\nTo run our pipeline on signal-level data (FAST5 files), we use the following command:\nnextflow run nf-core/viralrecon \\\n --input SAMPLESHEET_CSV \\\n --outdir OUTPUT_DIRECTORY \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version PRIMER_VERSION \\\n --skip_assembly \\\n --platform nanopore \\\n --fastq_dir fastq_pass/ \\\n --fast5_dir fast5_pass/ \\\n --sequencing_summary sequencing_summary.txt \\\n -profile conda,singularity,docker\nSome of the key options are:\n\n--platform nanopore makes sure that the correct sub-workflow will be used.\n--fastq_dir specifies the directory containing the FASTQ files generated by the guppy_basecaller program (this is the standard software from Nanopore that processes the raw signal data from the sequencing device). This directory should contain sub-directories for each barcoded sample following the naming convention barcodeXXXX (where X is a number between 0 and 9). By default, guppy_basecaller generates a folder called “fastq_pass” which follows this convention.\n--fast5_dir specifies the directory containing the FAST5 files generated by guppy_basecaller. This directory follows the same naming convention as above and is usually in a folder called “fast5_pass”.\n--sequencing_summary is a path to the “sequencing_summary.txt” text file generated by guppy_basecaller.\n\n\n\n\nApart from the specific options used by each sub-workflow, there are some general options that are used:\n\n--input specifies a CSV file with details about our samples. The format of this file depends on the specific sub-workflow you are using. See the details in the samplesheet documentation page.\n--outdir specifies the output directory to store all our results.\n--protocol amplicon sets the pipeline for PCR amplicon data (the other option is --protocol metagenomic, which we do not cover in this course).\n--genome 'MN908947.3' this is the standard name of the Wuhan-Hu-1 reference genome.\n--primer_set artic at the moment only “artic” primers are available by default. It is possible to use custom primers with the Illumina workflow (see details here).\n--primer_set_version the version of the ARTIC primer scheme used. The viralrecon primer config file indicates the available primer shemes are: 1, 2, 3, 4, 4.1, 5.3.2 and also 1200 (the 1200bp amplicon protocol, also known as “midnight”).\n--skip_assembly this is used to skip de-novo assembly of the genome. This step is unnecessary in amplicon protocols, which instead rely on mapping reads to the reference genome (reference-based assembly). De-novo assembly is necessary for metagenomic protocols.\n--skip_pangolin and --skip_nextclade is used to skip the lineage assignment. The reason is that viralrecon does not use the latest version of the SARS lineage databases, so we skip this step for now, and run it separately in a later step of the analysis.\n\nThere are two more generic options we used:\n\n-r 2.6.0 indicates the version of the pipeline we want to use. It’s always good to check what the latest version is on the viralrecon website. -profile singularity indicates how we want to manage the software required by the pipeline. In our case, we are using a software called Singularity, which creates a “virtual operating system” (called a container) where all the necessary software is run from. This ensures that all of the software is automatically installed and runs on any Linux computer.\n\n\n\n\n\n\n\nConda, Singularity, Docker?\n\n\n\nGenerally speaking, workflow management software such as Nextflow or Snakemake support three solutions for managing software dependencies:\n\nDocker is a software that allows to package a small virtual operating system (or a “container”) containing all the software and data necessary for running an analysis.\nSingularity also creates software containers, similarly to Docker. However, it can more easily interface with the user’s filesystem, without the need to have special permissions.\nConda is a package manager, also very popular in bioinformatics. Instead of creating virtual OS containers, Conda instead creates software environments (think of them as directories) where all the software is locally installed, including all its dependencies. The use of individual environments ensures that software packages with incompatible dependencies can still be used within the same pipeline.\n\nOf the three, Singularity is the recommended choice, although Conda is also a good alternative.\n\n\n\n4.3.1 Running the Workflow\nLet’s see an example in action by using some example data. If you go to the directory uk_illumina/ in the course materials, you will find several FASTQ files in the data directory. There is also a shell script (scripts/run_illumina_workflow.sh) that contains the commands we will use to run the workflow on these data.\nOpening the script, we can see the following commands:\n# create output directory\nmkdir results\n\n# run the workflow\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --platform illumina \\\n --input samplesheet.csv \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\nIt first creates a results directory (to store our output files) and then runs the nextflow command using the Illumina sub-workflow. We could run these commands one at a time by copy/pasting them to the terminal. Or alternatively, we can run the entire script using bash scripts/run_illumina_workflow.sh\nWhen you start running the workflow, you will get a list of the workflow steps and their progress. This may take quite a while to run, depending on the computer resources you have available. Once the workflow is complete, you should see a message similar to the following:\n-[nf-core/viralrecon] 8/8 samples passed Bowtie2 1000 mapped read threshold:\n 280038: GB16\n 2946614: GB28\n 252871: GB09\n 3269412: GB21\n 103742: GB23\n 3016474: GB36\n ..see pipeline reports for full list\n-\n-[nf-core/viralrecon] Pipeline completed successfully-\nCompleted at: 18-May-2022 08:08:25\nDuration : 1h 13m\nCPU hours : 8.1\nSucceeded : 343\nYou should also get several output files in the results folder specified with our nextflow command. We will detail what these files are in the next section.\n\n\n\n\n\n\nRunning the pipeline on our training computers\n\n\n\nOur training computers don’t have the high specifications needed for routine bioinformatic analysis, so the Illumina pipeline takes up to 1h to complete.\nWe provide already pre-processed results for 48 samples in the folder uk_illumina/preprocessed, which you can use to follow the next section."
+ },
+ {
+ "objectID": "materials/02-isolates/01-consensus.html#exercises",
+ "href": "materials/02-isolates/01-consensus.html#exercises",
+ "title": "4 Consensus assembly",
+ "section": "4.4 Exercises",
+ "text": "4.4 Exercises\n\n\n\n\n\n\nRunning nf-core/viralrecon: ONT data\n\n\n\n\n\n\n\nGo to the course materials directory india_nanopore. This contains Nanopore sequencing data for several samples collected in India. Nanopore data is organised in directories named as barcodeXX (where XX is a number) - this is the standard output from the Guppy software used to do basecalling and generate FASTQ files.\nThe Nanopore Medaka workflow expects to be given as an input a CSV file with two columns: sample name and barcode number. We already provide this file in samplesheet.csv.\nYour task now is to process these samples using the nf-core/viralrecon pipeline:\n\nUsing nano, open the script found in scripts/run_medaka_workflow.sh.\nFix the code in the script where you see the word “FIXME”:\n\nOutput the results to a directory called results/viralrecon/.\nThe input sample sheet is in the file samplesheet.csv (check the pipeline documentation to review what the format of this samplesheet should be for the Nanopore pipeline).\nThe FASTQ files are in a folder data/fastq_pass.\n\nRun the script using bash. This may take ~15 minutes to complete.\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nWe can open the script with Nano using the command:\nnano scripts/run_medaka_workflow.sh\nThe fixed code is:\n# create output directory\nmkdir -p results\n\n# run the workflow\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_cpus 8 --max_memory \"24.GB\" \\\n --input samplesheet.csv \\\n --outdir results/viralrecon \\\n --platform nanopore \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --skip_assembly \\\n --fastq_dir data/fastq_pass/ \\\n --artic_minion_caller medaka \\\n --artic_minion_medaka_model r941_min_high_g360\nWhat we did to fix the code was:\n\nSet --input as samplesheet.csv, which is the CSV file with two columns: sample name and Nanopore barcode number. The format of this file is detailed in the nf-core/viralrecon documentation.\nSet --outdir as results/viralrecon/, which is the directory where we want to output our results.\nSet --fastq_dir as data/fastq_pass, which is the directory containing the FASTQ file folders named as barcodeXX (where XX is a number). This is the standard output from the Guppy software used to do basecalling of Nanopore data to produce FASTQ files.\n\nWhen we finish editing the file we can hit Ctrl + X to exit Nano. Before it closes it will ask us if we want to save the file and we can type “Y” for yes.\nTo run the script we can do bash scripts/run_medaka_workflow.sh. After the workflow completes, we get a message similar to this one:\n-[nf-core/viralrecon] Pipeline completed successfully-\nCompleted at: 18-May-2022 08:08:25\nDuration : 13m 22s\nCPU hours : 1.6\nSucceeded : 167\nNote that the exact time that the workflow takes to run may differ from what we show here. The time depends on how many samples you are processing and how big the computer you are using is."
+ },
+ {
+ "objectID": "materials/02-isolates/01-consensus.html#summary",
+ "href": "materials/02-isolates/01-consensus.html#summary",
+ "title": "4 Consensus assembly",
+ "section": "4.5 Summary",
+ "text": "4.5 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nThe main steps to generate SARS-CoV-2 consensus sequences are: filter high-quality reads, map reads to reference genome, trim PCR primers and variant/mutation calling, which is finally used to produce a consensus sequence.\nOther steps that can be done include annotating the variants/mutations (what their effects in each gene might be) and assigning sequencences to known lineages/clades.\nNextflow is a software used for building workflows/pipelines involving multiple tools and data processing steps. Using established pipelines helps with automation, reproducibility, consistency of results and reduces the chances of data processing errors.\nThe nf-core/viralrecon pipeline implements the steps to generate SARS-CoV-2 consensus sequences from Illumina or Nanopore data.\nThe command nextflow run nf-core/viralrecon is used to run the pipeline, using specific options depending on the data we have."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#output-files",
+ "href": "materials/02-isolates/02-qc.html#output-files",
+ "title": "5 Quality Control",
+ "section": "5.1 Output Files",
+ "text": "5.1 Output Files\nAfter running our pipeline, we get several output directories and files. The directories we get depend on which version of the workflow was used (Illumina or Nanopore). The description of the output is detailed in the pipeline documentation. Although there are many output files, most of these contain results that are aggregated in an interactive MultiQC report, which makes their analysis easier. We highlight some of the main files of interest below.\n\nIlluminaNanopore (FASTQ)\n\n\n\nThe file multiqc/multiqc_report.html contains a MultiQC quality and analysis report for the consensus assemblies generated by the pipeline.\nThe folder variants/bowtie2/ contains individual BAM files, which can be visualised with IGV if we want to look at the reads mapped to the reference genome.\nThe folder variants/ivar/consensus/bcftools contains individual FASTA files (named *.consensus.fa) for each sample’s consensus genome sequence.\nThe file variants/ivar/variants_long_table.csv contains a table with the aggregated results of all the variants detected in each sample.\n\n\n\n\nThe file multiqc/medaka/multiqc_report.html contains a MultiQC quality and analysis report for the consensus assemblies generated by the pipeline.\nThe folder medaka/ contains:\n\nIndividual BAM files (named *.primertrimmed.rg.sorted.bam), which can be visualised with IGV if we want to look at the reads mapped to the reference genome.\nIndividual FASTA files (named *.consensus.fasta) for each sample’s consensus genome sequence.\nA file called variants_long_table.csv with a table of all the variants detected in each sample."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#sec-consensus-qc",
+ "href": "materials/02-isolates/02-qc.html#sec-consensus-qc",
+ "title": "5 Quality Control",
+ "section": "5.2 Quality Control",
+ "text": "5.2 Quality Control\nThe viralrecon pipeline produces many quality control metrics, which are conveniently compiled in an interactive report with MultiQC, as mentioned above. We will not detail here every section of the report (check the pipeline documentation for a full description), but only highlight some of the sections that can be used for a first assessment of the quality of our samples.\n\n5.2.1 Variant Calling Metrics\nThe first section or the report - Variant Calling Metrics - contains a summary table with several statistics for each sample, including the total number of reads, the number/percentage of reads mapped to the reference genome, the depth of coverage, the number of SNPs (single-nucleotide polymorphisms) and INDELs (insertions/deletions), the number of missense variants (mutations that result in an aminoacid change) and the fraction of ambiguous bases ‘N’ per 100kb.\nLooking at these basic metrics gives us a good first idea of whether some of the samples have a high fraction of missing bases (marked as N in the FASTA file), leading to a poorer assembly. We can quickly check this by sorting the table by the column named “# Ns per 100kb consensus” (you can convert the values in this column to a percentage by dividing the numbers by 100). There is also the ability to produce simple scatterplots from the data in this table, which can be useful to look at the relationship between the different metrics (see an example in the figure below).\nThis table also contains information about the lineage/clade assigned to each sample by the programs Pangolin and Nextclade. This gives us an idea of which samples may be more similar to each other, and where they fit in the global context of other sequences available publicly. We will talk more about this topic in the lineage assignment section.\n\n\n\n\nSnapshot of the “Variant Metrics” section of the viralrecon MultiQC report. Simple scatterplots can be made from the data on this table using the Plot button. For example, at the bottom we show a scatterplot showing the relationship between the median depth of coverage and the number of ambiguous bases ‘N’ per 100kb. For the data in this example, we can see that when the average depth of coverage drops below around 200 reads we start getting higher number of missing bases in the assembly.\n\n\n\n\n\n\n\n\nTerminology Alert!\n\n\n\nThe word coverage is sometimes used in an ambiguous way by bioinformaticians. It can mean two things:\n\nThe number of reads aligning to a position of the genome. This is sometimes called sequencing depth or depth of coverage (we will use these terms in the materials to avoid confusion). For example, we may say that the average depth of coverage of the genome is 20x, meaning that on average there are 20 reads aligned to a position of the genome.\nThe fraction of a genome that has a sufficient number of reads for analysis. For example, if 90% of the genome has a sufficient number of reads to be analysed (for example, at a threshold of 10x), we would say it has 90% coverage (we “covered” 90% of the genome with enough data). In the context of genome assembly, we sometimes use the word “coverage” to refer to the percentage of the consensus genome without ambiguous ‘N’ bases.\n\nIn the viralrecon MultiQC report the word “coverage” is used to mean “depth of coverage”.\n\n\n\n\n5.2.2 Amplicon Depth of Coverage\nThe next section of the report - Amplicon Coverage Heatmap - contains a graphical representation of the average depth of coverage for each PCR amplicon in the ARTIC protocol (i.e. the average number of reads mapped to each amplicon interval). This plot is extremely useful to identify common PCR dropouts occurring across many samples. This may be an indication that our PCR primer scheme is not working for some of the forms of the virus circulating in our population (for example due to mutations that accumulate in the primer site).\n\n\n\nExample heatmap showing the depth of coverage for a set of samples. The failure of an amplicon to be sequenced in several samples can be seen as a “column” of dark cells in the heatmap.\n\n\nWe can investigate primer dropout in more detail, for example by looking at the BAM files with mapped reads using IGV.\nFrom our heatmap (shown in the Figure above) we can see one of the PCR fragments - ncov-2019_83 - seems to have poor amplification across many of our samples. Let’s investigate this in more detail by looking at the alignment file:\n\nOpen IGV and go to File → Load from file….\nIn the file browser that opens go to the folder results/viralrecon/variants/bowtie2/ and select the file GB36.ivar_trim.sorted.bam to open it.\nGo back to File → Load from file… and this time load the BED files containing the primer locations. These can be found in resources/primers/artic_primers_pool1.bed and resources/primers/artic_primers_pool2.bed.\n\nOnce you have IGV open, you can navigate to the region where this amplicon is located by searching for the name of one of the primers - “ncov-2019_83_LEFT” - in the search box at the top. By zooming in to the region where this primer is located, we can see there is a mutation right at the start of this primer, which suggests that this may be the reason why this PCR fragment fails to amplify in this sample.\n\n\n\nScreenshot from the IGV program showing an example of a region with PCR amplicon dropout. This is shown by a lack of reads mapped to this region of the genome. Because the PCR fragments from the ARTIC protocol overlap between the two pools, we can see if there are mutations occurring at the primer sites. In this example, a mutation in the left primer of amplicon 83 suggests this may be the reason for the dropout in this sample."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#mutationvariant-analysis",
+ "href": "materials/02-isolates/02-qc.html#mutationvariant-analysis",
+ "title": "5 Quality Control",
+ "section": "5.3 Mutation/Variant Analysis",
+ "text": "5.3 Mutation/Variant Analysis\nOne of the output files produced by the pipeline is a table SNP and indel variants detected in our samples saved as a CSV file in variants/ivar/variants_long_table.csv. This table can be useful to investigate if particular mutations are particularly frequent in our samples and what their likely effects are.\n\n\n\nVariants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases.\n\n\nThe columns in this table are:\n\nSAMPLE is the name of our sequenced sample.\nCHROM is the name of the “chromosome”, which in our case is just the name of the reference genome (“MN908947.3”).\nPOS is the position of the genome where the mutation occurs.\nREF is the reference nucleotide.\nALT is the alternative nucleotide, that is the nucleotide that was observed in our sample.\nFILTER indicates whether the SNP passed quality filters from the variant calling software (“PASS”) or whether some other issue was observed (for example “dp” means that the depth of sequencing was unusually high or low).\nDP is the total depth of sequencing at this position, meaning how many reads in total were aligned there.\nREF_DP is the depth of sequencing of the reference allele, meaning how many reads contained the reference nucleotide.\nALT_DP is the depth of sequencing of the alternative allele, meaning how many reads contained the alternative nucleotide.\nAF is the allele frequence of the alternative allele, meaning the proportion of reads that contained the alternative nucleotide (this column is equivalent to ALT_DP/(ALT_DP + REF_DP)).\nGENE is the name of the gene in the SARS-CoV-2 annotation.\nEFFECT this is the predicted effect of the mutation in the gene. This output comes from the snpeff software and uses The Sequence Ontology nomenclature (follow the link to search for each term).\nHGVS_C, HGVS_P and HGVS_P_1LETTER is the DNA or amino acid change using HGVS nomenclature. For example, “c.1112C>T” means a C changed to a T at position 1112 of the genome; and “p.Pro371Leu” would mean that a Proline changed to a Leucine at position 371 of the respective protein.\nCALLER is the software used for variant calling.\nLINEAGE is the Pangolin lineage that the sample was assigned to. Note that this column should usually be ignored, since viralrecon doesn’t use the latest Pangolin data version by default.\n\nThis table can be opened in a spreadsheet program such as Excel to look at particular features. In terms of quality-control, we can filter the table:\n\nfor mutations with intermediate allele frequency (say < 70%) – if a sample has too many such mutations, that could indicate cross-contamination between samples.\nfor frameshift mutations – these mutations should be rare because they are highly disruptive to the functioning of the virus, and their occurrence is more often due to errors. The presence of these mutations are usually not critical for downstream analysis (such as lineage assignment and phylogenetics), but we should make a note that these mutations may be false-positives in our data.\n\nThis variants/mutations table can also be used to explore reasons for amplicon dropout. For example, we can identify how many samples contain the mutation we previously saw in the “ncov-2019_83_LEFT” primer (we saw this was in position 25,003 of the genome). We can do this by sorting our table by the column “POS” (position) and then scrolling down to find the samples with this mutation. We will find that only 3 of the samples contain this mutation, suggesting some other additional causes are leading to the dropout in this PCR fragment.\nFinally, for the Illumina pipeline (--platform illumina), it is important to note that not all of the mutations on this table are included in the final consensus. Only mutations with allele frequency >75% (AF >= 0.75) and minimum depth of 10 (DP >= 10) are retained in the final consensus. Therefore, to see which mutations are actually present in our consensus sequences, we need to filter this table for these criteria. Again, we note that this only applies to the Illumina pipeline. For the Nanopore pipeline (--platform nanopore) all the mutations included in this table are retained in the consensus sequence.\n\n\n\n\n\n\nORF1b Annotation Issues\n\n\n\nThe annotation of the ORF1b gene had some changes between the first original assembly and newer versions of this annotation. In particular, there is a frameshift that is not considered in the annotation used by most software, and this causes a discrepancy in the results between viralrecon and other tools such as Nextclade (which we will cover in the next section).\nThis issue is under investigation by the developers of viralrecon and may be corrected in future versions of the pipeline.\nFor now, the advice is to ignore the variant effects of ORF1b as these correspond to an older version of the annotated gene.\n\n\n\n\n\n\n\n\nKeep Original Files Intact\n\n\n\nWhen you analyse the results of a CSV file in a spreadsheet software (such as Excel), it may be a good idea to create a copy of your file to avoid accidentally modifying the original table. For example, if you accidentally sort one column of the table but forget to sort other columns, of if you accidentally filter the results and delete some of the rows from the table, etc.\nYou can save a copy of the file by going to File → Save As…. You may want to save the copy as an Excel file format, to include graphs and colours. (Remember that the CSV format is a plain text file - it does not support graphs or coloured cells.)"
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#cleaning-fasta-files-optional",
+ "href": "materials/02-isolates/02-qc.html#cleaning-fasta-files-optional",
+ "title": "5 Quality Control",
+ "section": "5.4 Cleaning FASTA Files (Optional)",
+ "text": "5.4 Cleaning FASTA Files (Optional)\nTo proceed with our analysis, we need a FASTA file containing all of our consensus sequences. However, our viralrecon pipeline outputs separate FASTA files for each sample. We can see this by running (from within the uk_illumina/ directory):\nls results/viralrecon/variants/ivar/consensus/bcftools/\nAlso, the workflow modifies our original sample names in the FASTA file, by adding extra information to the sequence name. For example:\nhead -n 1 results/viralrecon/variants/ivar/consensus/bcftools/ERR5728910.consensus.fa\n>ERR5728910 MN908947.3\nWhat we want to do is clean these sample names, so that we end up with:\n>ERR5728910\nWe also want to make sure to combine all the samples into a single FASTA file.\nWe can the command cat to combine (concatenate) the individual files and the sed command to substitute text and clean our sample names. Let’s do this step by step.\nFirst, we can use the * wildcard to combine all the FASTA files with the cat command:\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.fa\nRunning this command will print all of the sequences on the screen! To see what happened a little better, we could pipe this command to less to browse up-and-down through the file:\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.fa | less\nWe could also check that we now have all our samples combined, we could pass the results to grep and search for the word >, which in the FASTA format indicates the sequence name:\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.fa | grep \">\" | wc -l\nThis should give us 7 as the result (which makes sense, since we have 7 samples).\nWe can now proceed with cleaning the names of the sequences, by using sed:\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.fa | sed 's| MN908947.3||' > results/clean_sequences.fa\nNotice that in this last command we make sure to redirect the result to a new file using >."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#missing-data-intervals",
+ "href": "materials/02-isolates/02-qc.html#missing-data-intervals",
+ "title": "5 Quality Control",
+ "section": "5.5 Missing data intervals",
+ "text": "5.5 Missing data intervals\nWhen we align reads to the reference genome, there may be regions that we were not able to sequence (e.g. due to amplicon dropout, discussed above) and therefore cannot tell what the base sequence in those positions is. In those cases, the viralrecon pipeline includes the missing character ‘N’ in the sequence.\nAs a further quality check, it is useful to check how many contiguous intervals of missing bases we have in each of our assemblies, as well as how long those intervals are. For example, for this small sequence:\nT A N N N G C T N N A T\nWe have two missing intervals, from positions 3-5 and from positions 9-10.\nTo obtain a list of missing intervals in a sequence, we can use the software seqkit, which is a toolkit of commands to do many operations on FASTA/FASTQ files (check out its documentation). In particular, we can use the following command:\nseqkit locate -i -P -G -M -r -p \"N+\" results/clean_sequences.fa\nseqID patternName pattern strand start end\nGB09 N+ N+ + 1 342\nGB09 N+ N+ + 9246 9502\nGB09 N+ N+ + 10738 11331\nGB09 N+ N+ + 21428 21543\nGB09 N+ N+ + 27400 27462\nGB09 N+ N+ + 27618 27644\nGB09 N+ N+ + 29827 29893\n\n... more output omitted...\nThe output is a tabular file specifying the consensus sequence name (first column) and the location of intervals in the genome where the pattern of one or more “N” were found.\nTo see what all the options we used with this command are, see the tool’s documentation."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#exercises",
+ "href": "materials/02-isolates/02-qc.html#exercises",
+ "title": "5 Quality Control",
+ "section": "5.6 Exercises",
+ "text": "5.6 Exercises\n\n\n\n\n\n\nQuality control for India (ONT) samples\n\n\n\n\n\n\n\nNow it’s your turn to check the quality of the assembled genomes from our India samples, sequenced using Nanopore. For this exercise, we will use the results produced by viralrecon on 48 pre-processed samples:\n\nUse the file explorer to open the folder preprocessed, and then:\n\nOpen the file in pipeline_info/execution_report_DATE.html (where “DATE” is the date when the files were processed).\nHow long did the workflow take to run?\nWhich step of the pipeline took the longest to run?\n\nOpen the MultiQC report found in preprocessed/multiqc/medaka/multiqc_report.html and answer the following questions:\n\nProduce a plot between median depth of coverage (x-axis) and #Ns (y-axis). What do you think is the average coverage needed for <10% of missing data.\nWere there any PCR amplicons with a dropout (i.e. very low depth of coverage) across multiple samples?\nDo any of these dropout amplicons coincide with gene “S” (Spike protein gene)? Note: you can see the ARTIC PCR primer location from the online repository and the gene locations in the SARS-CoV-2 NCBI genome browser.\n\nBased on any amplicons that you identified in the previous question, find if there are any mutations coinciding with their primer locations. You can use the mutation variant table in preprocessed/medaka/variants_long_table.csv.\nWith the mutation variants table open in a spreadsheet program (LibreOffice on our training machines):\n\nUse the filter button to look for mutations in gene “S” (encodes for the Spike protein).\nIdentify the samples and positions of mutations of the type “disruptive_inframe_deletion”.\nUsing IGV, open the BAM file for one of those samples. Note: BAM files are located in preprocessed/medaka/SAMPLE.primertrimmed.rg.sorted.bam (were ‘SAMPLE’ is the sample name).\nGo to the location of those mutations. From the read alignment, how confident are you about it? What could you do to confirm it?\n\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nQuestion 1\nWe open the file found in preprocessed/pipeline_info/execution_report_2022-05-04_12-41-12.html, which contains information about the pipeline that was run on a set of 48 samples. We can see at the top of the report that it took ~15 minutes to run. This report also gives us information about how long each individual step of the pipeline took the longest to run (and other information such as which used more CPU or RAM memory). For example, in the section “Job Duration” we can see a graph that looks like this:\n\nThis indicates that the step running the artic minion tool takes the longest. This is not surprising as this is the step where most of the work is happening (mapping, primer trimming and making a consensus sequence). You can revise the steps of the workflow in the respective section above.\n\nQuestion 2\nWhen opening the MultiQC report, we can see the first section contains a table with several statistics about the quality of each sample. We can produce a plot from this table, by pressing the “Plot” button above the table. The plot we were asked for is the following:\n\n\n\nThis plot can be produced by clicking the Plot button on top of the table and then selecting the variables “Coverage median” and “# Ns per 100kb consensus”.\n\n\nThe y-axis of the plot tells us the number of ‘N’ per 100kb of sequence. If we divide those numbers by 100 we get a percentage and from these samples it seems like a median depth of coverage of around 200 reads generates samples with less than 10% of missing bases.\nFrom the section of the report “Amplicon coverage heatmap” we can see the average depth of coverage for each PCR amplicon from the ARTIC protocol. There are 3 amplicons in particular that have very low depth of coverage: nCoV-2019_51, nCoV-2019_62 and nCoV-2019_95.\nWe can check the ARTIC V3 primer BED file online to look at the location of these primers, or we could look for it using command-line tools. Here is an example:\ncat resources/primers/artic_version3_pool*.bed | grep \"_51_\"\nMN908947.3 15171 15193 nCoV-2019_51_LEFT 1 +\nMN908947.3 15538 15560 nCoV-2019_51_RIGHT 1 -\nFor this example, we can see the amplicon is between positions 15171 - 15560 of the genome. Looking at the SARS-CoV-2 NCBI genome browser, we can see this coincides with ORF1ab. If we do the same analysis for the other primers, we will see that none of them coincides with gene S.\n\nQuestion 3\nTo investigate whether this amplicon dropout is due to mutations in the primer regions we can open the mutations table preprocessed/medaka/variants_long_table.csv. If we sort the table by the column “POS”, we can scroll to the location of each primer pair.\nWe can see that in only one case there is a mutation coinciding one of the primers: in sample “IN33” position 28687 there is a C > T mutation, which coincides with primer nCoV-2019_95_LEFT. However, this only occurs for one of the samples, suggesting the reason for the PCR dropout might be related to other factors.\n\nQuestion 4\nAfter filtering our table for gene S (Spike gene), we can see only a couple of mutations of type “disruptive_inframe_deletion” in some of the samples at positions 21764 and 21990. To look at the reads with this mutation, we open the BAM file for one of these samples in IGV (for example sample IN22):\n\nGo to File → Load from file….\nIn the file browser that opens go to the folder preprocessed/medaka and select the file IN22.primertrimmed.rg.sorted.bam to open it.\nIn the search box we can type “NC_045512.2:21990” which will zoom-in on the region around the deletion.\n\nFrom looking at the reads aligned to this position of the genome, we can see several of them containing a 3bp deletion. However, we can also see some reads that do not contain the deletion, and others that seem to be mis-aligned. If we were interested in confirming this mutation, we could do an independent PCR followed by Sanger sequencing, for example.\nAlthough this was not part of the question, it is also worth noting that the primer “nCoV-2019_73_LEFT” starts very near this deletion. In the MultiQC report, we can see that this PCR amplicon had very poor amplification in this sample. One possibility is that the deletion interfered with the primer efficiency in this sample.\nGenerally, we don’t need to confirm every single mutation obtained from our analysis. But if we see a mutation occurring many times, then it may be worth further investigation, especially if it is disruptive and in an important gene such as the Spike gene.\n\n\n\n\n\n\n\n\n\n\n\n\n\n\n\n\nClean FASTA file: ONT data\n\n\n\n\n\n\n\nIn this exercise we will create a clean FASTA file for the samples collected in India.\nIn this case, the output FASTA files are in the folder results/viralrecon/medaka and have the file extension .fasta. If we look at one of the files:\nhead -n 1 results/viralrecon/medaka/IN42.consensus.fasta\n>IN42/ARTIC/medaka MN908947.3\nWe can see that the name has a lot of extra information attached to it. We want to clean the name of the sequences so that the result is:\n>IN42\nThe following sed command can be used to substitute the text “/ARTIC/medaka MN908947.3” with nothing:\nsed 's|/ARTIC/medaka MN908947.3||'\n\nPipe the tools cat and sed to construct a command that generates a new file called results/clean_sequences.fa containing all the sequences with “clean” sequence names.\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nThe complete code to achieve the desired outcome is:\ncat results/viralrecon/medaka/*.consensus.fasta | sed 's|/ARTIC/medaka MN908947.3||' > results/clean_sequences.fa\nLook at our companion Unix course materials for more information about how the sed command works."
+ },
+ {
+ "objectID": "materials/02-isolates/02-qc.html#summary",
+ "href": "materials/02-isolates/02-qc.html#summary",
+ "title": "5 Quality Control",
+ "section": "5.7 Summary",
+ "text": "5.7 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nThe output of the pipeline includes, among others:\n\nA detailed MultiQC report, including information about genome coverage, the depth of sequencing for each amplicon, as well as other quality metrics that can help to troubleshoot problematic samples.\nA table with SNP and indel mutation variants identified in each sample.\nInformation about SARS-CoV-2 lineages/clades/variants, which is detailed in the next section."
+ },
+ {
+ "objectID": "materials/02-isolates/03-lineages.html#sec-lineages",
+ "href": "materials/02-isolates/03-lineages.html#sec-lineages",
+ "title": "6 Lineages and variants",
+ "section": "6.1 SARS-CoV-2 Variants",
+ "text": "6.1 SARS-CoV-2 Variants\nAs viruses (or any other organism) evolve, random DNA changes occur in the population, for example due to replication errors. Many of these changes are likely to be neutral, meaning that they do not change the characteristics of the virus in any significant way. Neutral changes tend to drift in the population, increasing or decreasing in frequency in a random way, but most likely end up disappearing due to their low starting frequency in the population.\nOn the other hand, advantageous mutations can occur, which lead to changes in the characteristics of the virus that are beneficial for its spread in the population (e.g. high transmissibility, resistance to immune response or vaccines, etc.). Such beneficial mutations will therefore experience positive selection, potentially leading to their increase in frequency in the population as they spread to more and more hosts.\nViruses carrying those advantageous mutations may, over time, aquire other advantageous mutations that further increase their fitness and, therefore, their frequency in the population. One way to visualise this is by looking at a phylogenetic tree showing the relationship between sequences (based on their similarity) and how groups of sequences change over time.\n\n\n\nExample of global phylogeny from the Nextstrain public server. Colours show different Nextstrain clades. (Screenshot taken Feb 2022)\n\n\nIn the figure above, which shows SARS-CoV-2 samples from across the world, we can see groups of similar sequences rapidly “expanding” at certain points in time. Such groups of sequences, which share a collection of DNA changes, are referred to as SARS-CoV-2 variants (see box below about the ambiguous meaning of this term). In an effort to understand the spread of the virus and monitor situations of increased occurrence of such variants, several groups and institutions have developed a system to classify groups of SARS-CoV-2 sequences as variants of interest and variants of concern.\nA full explanation and definitions of such variants is given in the World Health Organisation (WHO) variants page The main classification systems currently in use are:\n\nGISAID clades\nNextstrain clades\nPango lineages\nWorld Health Organisation (WHO) variants\n\nIn practice, there is a big overlap between these different nomenclature systems, with WHO variants having a direct match to Pango lineages and Nextstrain clades. In fact, the different teams work together to try and harmonise the nomenclature used, and thus facilitate the interpretation of sequence analysis.\nThe two most popular systems - Nextclade and Pangolin - have slightly different levels of resolution. Nextclade’s nomenclature system was developed to highlight diversity patterns at a larger scale, allowing discussions of SARS-CoV-2 diversity at a global level and over larger time scales. On the other hand, Pangolin’s nomenclature system is more fine-grained, aiming to follow the dynamics of the pandemic as it unfolds. The two systems are complementary to each other, and our analysis of SARS-CoV-2 sequences should include both tools.\n\n\n\n\n\n\nWhat is a variant?\n\n\n\nIt is important to note that the term variant can be sometimes ambiguous.\nThe term “SARS-CoV-2 variant” usually refers to the WHO definition of variants of concern/interest (e.g. the Alpha, Delta and Omicron variants), which includes sequences containing a collection of several nucleotide changes that characterise that group. According to this definition, we have two variants in the example below (samples 1 & 2 are one variant and samples 3 & 4 another variant).\n\nHowever, in bioinformatic sequence analysis, a sequence variant refers to an individual change in the DNA sequence (a SNP or an insertion/deletion). Using this definition, in the example above we have 5 variants: 3 SNPs and 2 indels. In the Consensus Sequence section, we saw that one of our workflow steps was “variant calling”. This was the definition of variant we were using: identifying individual SNPs and/or indels relative to the reference genome, from our sequencing data. This is also reflected in the one of the common file formats used to store SNP/indel information, the VCF file, which means “Variant Call Format”.\nSometimes the term “mutation” is used to refer to SNP/indel variants. For example see this definition from the COG consortium.\nBecause of this ambiguity, the terms “lineages” or “clades” are often used instead of “variants” when referring to groups of similar SARS-CoV-2 sequences, because they have a phylogenetic interpretation."
+ },
+ {
+ "objectID": "materials/02-isolates/03-lineages.html#pangolin",
+ "href": "materials/02-isolates/03-lineages.html#pangolin",
+ "title": "6 Lineages and variants",
+ "section": "6.2 Pangolin",
+ "text": "6.2 Pangolin\n\nThe first tool we will cover is called pangolin and uses the Pango nomenclature system. The main steps performed by this tool are:\n\nMultiple sequence alignment of our samples against the Wuhan-Hu-1 reference genome, using the minimap2 software.\nAssigning our sequences to lineages based on the current global phylogeny. Two methods/software are available:\n\npangoLEARN (default) uses a pre-trained machine learning model.\nUShER uses a more classic parsimony-based method, but highly optimised for working with large numbers of sequences.\n\nClassifying our sequences according to the WHO nomenclature of variants of interest/concern using the scorpio software.\n\nAlthough Pangolin can run as part of the nf-core/viralrecon pipeline we used, we recommended to turn this option off. The reason is that the model to classify variants regularly changes over time, as more public sequences become available and the nomenclature rules updated. Therefore, it important to always run the samples through the latest Pangolin version available.\nPangolin can be run from the command line, using two steps:\n\nUpdating the data used for lineage/variant classification.\nRunning the actual lineage assignment step.\n\nOn our example data, these would be the commands:\n# update pangolin data\npangolin --update-data\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv results/clean_sequences.fa\n\nThe first command downloads the latest version of the lineages and their characteristic mutations from the Pango classification system.\nThe second command runs the FASTA file of consensus genomes through Pangolin’s classification algorithm.\n\n--outdir is used to define the directory to save the results in.\n--outfile is used to give the file a name of our choice.\n\n\nThe output is a CSV file, with several columns of interest, including WHO variants of concern identified using the Scorpio software. A detailed explanation of the columns of this file is given in the Pangolin documentation page.\nMore information about running Pangolin from the command line can be found in its online documentation.\n\n6.2.1 Web Application\nThis tool can also be run separately using a web application, which only requires us to provide with a FASTA file of consensus sequences. This may desirable to re-run samples using the latest version of the Pangolin software and SARS-CoV-2 variant databases.\nThe results from the web application can be downloaded as a CSV file, which contains a table similar to the one obtained from our command above (some of the column names are different, but their meaning is the same)."
+ },
+ {
+ "objectID": "materials/02-isolates/03-lineages.html#nextclade",
+ "href": "materials/02-isolates/03-lineages.html#nextclade",
+ "title": "6 Lineages and variants",
+ "section": "6.3 Nextclade",
+ "text": "6.3 Nextclade\n\nAnother system of clade assignment is provided by nextclade, which is part of the broader software ecosystem Nextstrain.\nNextclade performs similar steps to Pangolin, with some differences in the algorithms that are used:\n\nEach sequence is aligned with the Wuhan-Hu-1 reference genome using a local alignment algorithm.\nSamples are placed in the global phylogeny using a distance-based metric (placing the sequence on the tree where it has the highest similarity with).\nClade assignment is done based on the previous phylogeny placement step.\n\nYou can find more details about Nextclade’s methods on its documentation. Nextclade also provides several quality control metrics, which are very useful to identify problematic samples.\nAs we discussed above, the models and clades are regularly updated, so we also skipped this step when we ran the nf-core/viralrecon pipeline. Instead, we can run this tool directly from the command line, by first making sure to download the most up-to-date clade information. Here are the commands:\n# get nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade results/clean_consensus.fa\n\nThe first command (nextclade dataset get) downloads the latest version of the Nextclade dataset for SARS-CoV-2 (option --name sars-cov-2). We define the directory to store this information with --output-dir.\nThe next step is to run the actual clade assignment on our data. We use the database from the previous step as --input-dataset, we define a directory to output all the results with --output-all and at the end of the command we give as input our clean FASTA consensus genomes.\n\nMore information about running Nextclade from the command line can be found in its online documentation.\n\n6.3.1 Web Application\nNextclade offers an interactive application, which can be used to run its analysis on a FASTA file with sequences:\n\nGo to nextclade.org.\nClick Select a file to browse your computer and upload the FASTA file with the cleaned consensus sequences (results/clean_sequences.fa).\nNextclade will automatically detect your data are from SARS-CoV-2, but if not you can select this organism.\nClick Run.\n\nNextclade will show a progress of its analysis at the top of the page, and the results of several quality control metrics in the main panel (see Figure).\n\n\n\nOverview of the Nextclade web interface.\n\n\n\n\n\n\n\n\nNextclade and data privacy\n\n\n\nWhen using the Nextclade web application, the data does not leave your computer, so privacy concerns are not an issue."
+ },
+ {
+ "objectID": "materials/02-isolates/03-lineages.html#exercises",
+ "href": "materials/02-isolates/03-lineages.html#exercises",
+ "title": "6 Lineages and variants",
+ "section": "6.4 Exercises",
+ "text": "6.4 Exercises\n\n\n\n\n\n\nNextclade\n\n\n\n\n\n\n\nIn this exercise we will work with 48 consensus sequences from the UK, processed with the nf-core/viralrecon pipeline and covered in the previous section.\nGo to nextclade.org and load the sequences provided in uk_illumina/preprocessed/clean_sequences.fa.\n\nAre there any samples that were classified as “bad” quality? If so, what is the main reason?\nSort the table by the “Clade” column. Looking at the mutations in gene S on the right, you can see that all sequences classified as “Alpha” have a deletion in positions 21992-21994. Samples classified as “Delta” do not have this deletion and instead have a deletion in positions 22029-22034. However, there is one exception: sample GB39, classified as “Delta” has both deletions. Investigate if this mutation is accurate using IGV:\n\nOpen the BAM alignment file for this sample (the alignment file is in results/viralrecon/variants/bowtie2/GB39.ivar_trim.sorted.bam).\nOpen the BAM alignment file for one of the “Alpha” samples as a comparison.\nOpen the ARTIC primer files (two BED files found in resources/primers/).\nGo to the position where this deletion was identified and investigate if it seems clear from the mapped reads.\n\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nQuestion 1\nAfter loading the samples to the Nextclade web application, we can see that, generally, these samples have high quality as very few are highlighted by Nextclade’s analysis.\nSorting the table by the “QC” column, we can see two samples with bad quality (red) and one with mediocre quality (yellow).\nThere are separate reasons for the two bad quality samples:\n\nSample GB16 has low quality due to a high number of missing bases.\nSample GB39 has low quality due to the occurrence of too many “private mutations”. These are mutations that are only found in this sample, compared to other samples present in Nextclade’s background sequence tree. As we don’t usually expect too many new mutations to occur in a new sample (the mutation rate of SARS-CoV-2 is around 2 new mutations per month), the occurrence of too many private mutations could indicate sequencing quality issues.\n\nQuestion 2\nSort the table by the “Clade” column. Looking at the mutations in gene S on the right, you can see that all sequences classified as “Alpha” have a deletion in positions 21992-21994. Samples classified as “Delta” do not have this deletion and instead have a deletion in positions 22029-22034. However, there is one exception: sample GB39, classified as “Delta” has both deletions. Investigate if this mutation is accurate using IGV:\n\nOpen the BAM alignment file for this sample (the alignment file is in preprocessed/variants/bowtie2/GB39.ivar_trim.sorted.bam).\nOpen the BAM alignment file for one of the “Alpha” samples as a comparison.\nOpen the ARTIC primer files (two BED files found in resources/primers/).\nGo to the position where this deletion was identified and investigate if it seems clear from the mapped reads.\n\nBy sorting the table by “Clade”, we can see that although sample GB39 was classified as a Delta variant, it has two deletions in gene S that are present in Alpha variants, as shown in this snapshot (click the image to view a bigger size):\n\n \n\nAs we investigate one of these in more detail from the BAM file (opening it in IGV), we can see that there is an inconsistency between reads coming from the PCR fragment “nCoV-2019_72” and those reads coming from the “nCoV-2019_73” fragment. In some of those reads the deletion is present, but in others it is not. If we look at an Alpha variant sample (for example GB43) we can see that this deletion is present in both cases.\n\nIf we thought this sample was crucial for public health investigation, then this would require further investigation by doing a PCR with new primers and Sanger-sequencing the fragment, for example."
+ },
+ {
+ "objectID": "materials/02-isolates/03-lineages.html#summary",
+ "href": "materials/02-isolates/03-lineages.html#summary",
+ "title": "6 Lineages and variants",
+ "section": "6.5 Summary",
+ "text": "6.5 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nGroups of similar SARS-CoV-2 sequences are classified into lineages or clades by different groups. The main nomenclature systems in use are Nextstrain, Pangolin and GISAID.\nIn addition, the World Health Organisation (WHO) classifies some forms of SARS-CoV-2 as variants of concern or variants of interest. These are forms of the virus that have been determined to have a significant public health impact.\nBoth Pangolin and Nextclade assign consensus sequences to lineages/clades and additionally identify those that correspond to WHO variants of concern. Both of these are run as part of the nf-core/viralrecon pipeline, but can also be run using a web application:\n\nPangolin web application\nNextclade web application\n\nBesides clade assignment and variant classification, Nextclade provides additional analysis such as identification of mutations and quality control metrics that can be used to identify problematic samples."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#sec-phylogeny",
+ "href": "materials/02-isolates/04-phylogeny.html#sec-phylogeny",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.1 SARS-CoV-2 Phylogeny",
+ "text": "7.1 SARS-CoV-2 Phylogeny\n\nBuilding phylogenetic trees for SARS-CoV-2 is challenging due to the large number of sequences available, with millions of genomes submited to GISAID to this day. This means that using maximum-likelihood inference tools to build a global SARS-CoV-2 phylogeny is both time-consuming and computationally demanding.\nHowever, several researchers have dedicated their time to identifying tools and models suitable for the phylogenetic analysis of this organism. For example, the global phylogenies repository from Rob Lanfear provide with several tips on building phylogenetic trees for this organism. Their trees are regularly updated and available to download from the GISAID website (which requires an account).\nGlobal phylogenies are also available from the groups of Russell Corbett-Detig and Yatish Turakhia, who have developed efficient methods and tools for dealing with large phylogenies. These tools include UShER and matUtils, introducing a new and efficient file format for storing phylogenetic trees, mutations and other annotations (such as lineages) called mutation-annotated trees (MAT format). Their phylogenies are updated daily and are publicly available for download. (Note: Course materials covering these tools are still under development.)\nTwo popular tools used for phylogenetic inference via maximum-likelihood are FastTree and IQ-Tree. Generally, when building a tree from a collection of samples you can include the Wuhan-Hu-1 reference sequence as an outgroup to root the tree. Optionally, you can also add a collection of sequences from around the world (and across time) to contextualise your samples in the global diversity.\nAs an input, these programs need a multiple sequence alignment FASTA file, which is where we start our analysis.\n\n\n\n\n\n\nData for this section\n\n\n\nWe will work from the course materials folder called 04-phylogeny, which contains the following files:\n\ndata/uk_consensus.fa and data/india_consensus.fa are consensus sequences from the UK and India, respectively. These are the sequences previously assembled using the nf-core/viralrecon pipeline.\nsample_annotation.tsv is a tab-separated values (TSV) file with information about each sample such as the date of collection and lineage/clade they were assiged to from our analysis. We will use this table to annotate our phylogenetic trees. This table can also be opened in a spreadsheet program such as Excel.\nresources/reference/sarscov2.fa is the reference genome."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#sec-mafft",
+ "href": "materials/02-isolates/04-phylogeny.html#sec-mafft",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.2 Alignment",
+ "text": "7.2 Alignment\nThe first step in building a phylogenetic tree is to produce a multiple sequence alignment from all our consensus sequences. This is the basis for building a phylogenetic tree from the positions that are variable across samples.\nA widely used multiple sequence alignment software is called MAFFT. For SARS-CoV-2, a reference-based alignment approach is often used, which is suitable for closely-related genomes. MAFF provides this functionality, which is detailed in its documentation.\nWe demonstrate the analysis using the UK samples in our course materials. First, start by creating a directory for the output:\nmkdir -p results/mafft\nThen, we run the command to generate a reference-based alignment:\nmafft --6merpair --maxambiguous 0.2 --addfragments data/uk_consensus.fa resources/reference/sarscov2.fa > results/mafft/uk_alignment.fa\nThe meaning of the options used is:\n\n--6merpair is a fast method for estimating the distances between sequences, based on the number of short 6bp sequences shared between each pair of sequences. This is less accurate than other options available (like --localpair and --globalpair), but runs much faster in whole genome data like we have.\n--maxambiguous 0.2 automatically removes samples with more than 20% ambiguous ‘N’ bases (or any other value of our choice). This is a convenient way to remove samples with poor genome coverage from our analysis.\n--addfragments data/consensus_sequences.fa is the FASTA file with the sequences we want to align.\nfinally, at the end of the command we give the reference genome as the input sequence to align our sequences against.\n\nMAFFT provides several other methods for alignment, with tradeoffs between speed and accuracy. You can look at the full documentation using mafft --man (mafft --help will give a shorter description of the main options). For example, from its documentation we can see that the most precise alignment can be obtained with the options --localpair --maxiterate 1000. However, this is quite slow and may not be feasible for whole genome data of SARS-CoV-2.\n\n7.2.1 Visualising alignments\nWe can visualise our alignment using the software AliView, which is both lightweight and fast, making it ideal for large alignments. Visualising the alignment can be useful for example to identify regions with missing data (more about this below).\n\n\n\nSnapshop of an alignment visualised with AliView. In this case we are looking at the end of the alignment of our sequences, which shows a typical high number of missing (‘N’) bases.\n\n\n\n\n\n\n\n\nOther Alignment Strategies\n\n\n\nThere are other commonly used alignment tools used for SARS-CoV-2 genomes:\n\nThe minimap2 software has been designed for aligning long sequences to a reference genome. It can therefore be used to align each consensus sequence to the Wuhan-Hu-1 genome. This is the tool internally used by Pangolin.\nNextclade uses an internal alignment algorithm where each consensus sequence is aligned with the reference genome. The alignment produced from this tool can also be used for phylogenetic inference.\n\nIt is worth mentioning that when doing reference-based alignment, insertions relative to the reference genome are not considered."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#sec-iqtree",
+ "href": "materials/02-isolates/04-phylogeny.html#sec-iqtree",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.3 Tree Inference: IQ-Tree",
+ "text": "7.3 Tree Inference: IQ-Tree\nIQ-TREE supports many substitution models, including models with rate heterogeneity across sites.\nLet’s start by creating an output directory for our results:\nmkdir -p results/iqtree\nAnd then run the program with default options (we set --prefix to ensure output files go to the directory we just created and are named “uk”):\niqtree2 -s results/mafft/uk_alignment.fa --prefix results/iqtree/uk\nWithout specifying any options, iqtree2 uses ModelFinder to find the substituion model that maximizes the likelihood of the data, while at the same time taking into account the complexity of each model (using information criteria metrics commonly used to assess statistical models).\nFrom the information printed on the console after running the command, we can see that the chosen model for our alignment was “GTR+F+I”, a generalised time reversible (GTR) substitution model. This model requires an estimate of each base frequency in the population of samples, which in this case is estimated by simply counting the frequencies of each base from the alignment (this is indicated by “+F” in the model name). Finally, the model includes rate heterogeneity across sites, allowing for a proportion of invariant sites (indicated by “+I” in the model name). This makes sense, since we know that there are a lot of positions in the genome where there is no variation in our samples.\nWe can look at the output folder (specified with --prefix) where we see several files with the following extension:\n\n.iqtree - a text file containing a report of the IQ-Tree run, including a representation of the tree in text format.\n.treefile - the estimated tree in NEWICK format. We can use this file with other programs, such as FigTree, to visualise our tree.\n.log - the log file containing the messages that were also printed on the screen.\n.bionj - the initial tree estimated by neighbour joining (NEWICK format).\n.mldist - the maximum likelihood distances between every pair of sequences.\nckp.gz - this is a “checkpoint” file, which IQ-Tree uses to resume a run in case it was interrupted (e.g. if you are estimating very large trees and your job fails half-way through).\n.model.gz - this is also a “checkpoint” file for the model testing step.\n\nThe main files of interest are the report file (.iqtree) and the NEWICK tree file (.treefile).\n\n\n\n\n\n\nInference of very large trees\n\n\n\nAlthough running IQ-Tree with default options is fine for most applications, there will be some bottlenecks once the number of samples becomes too large. In particular, the ModelFinder step may be very slow and so it’s best to set a model of our choice based on other people’s work. For example, work by Rob Lanfear suggests that models such as “GTR+G” and “GTR+I” are suitable for SARS-CoV-2. We can specify the model used by iqtree2 by adding the option -m GTR+G, for example.\nFor very large trees (over 10k or 100k samples), using an alternative method to place samples in an existing phylogeny may be more adequate. UShER is a popular tool that can be used to this end. It uses a parsimony-based method, which tends to perform well for SARS-CoV-2 phylogenies."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#sec-figtree",
+ "href": "materials/02-isolates/04-phylogeny.html#sec-figtree",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.4 Visualising Trees",
+ "text": "7.4 Visualising Trees\nThere are many programs that can be used to visualise phylogenetic trees. In this course we will use FigTree, which has a simple graphical user interface.\nTo open the tree, go to File > Open… and browse to the folder with the IQ-Tree output files. Select the file with .treefile extension and click Open. You will be presented with a visual representation of the tree.\nWe can also import a “tab-separated values” (TSV) file with annotations to add to the tree. For example, we can use our results from Pangolin and Nextclade, as well as other metadata to improve our visualisation (we have prepared a TSV with this information combined, which you could do using Excel or another spreadsheet software).\n\nGo to File > Import annotations… and open the annotation file.\nOn the menu on the left, click Tip Labels and under “Display” choose one of the fields of our metadata table. For example, you can display the lineage assigned by Pangolin (“pango_lineage” column of our annotation table).\n\nThere are many ways to further configure the tree, including highlighting clades in the tree, and change the labels. See the figure below for an example.\n\n\n\nAnnotated phylogenetic tree obtained with FigTree. We used the lineage of each sample as our tip labels and aligned the labels on the right (check the tickbox at the top of the left menu called “Align tip labels”). We identified two clades in the tree that corresponded to the Alpha and Delta variants, and used the “Highlight” tool to give them different colours. To do this, change the “Selection Mode” at the top to “Clade”, then select the branch at the base of the clade you want to highlight, and press the “Highlight” button on the top to pick a colour."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#time-scaled-phylogenies",
+ "href": "materials/02-isolates/04-phylogeny.html#time-scaled-phylogenies",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.5 Time-scaled Phylogenies",
+ "text": "7.5 Time-scaled Phylogenies\nThe trees that we build from sequence data are scaled using the mutation rate estimated from the sequence alignments. This is useful if we want to know, for example, on average how many mutations separate different branches of the tree.\nAnother way to scale trees is to use time. For viral genome sequences, we usually have information about their date of collection, and this can be used to scale the phylogeny using the date information. The idea is to rescale the trees such that the x-axis of the tree represents a date rather than number of mutations.\nTwo programs that can be used to time-scale trees using date information are called TreeTime and Chronumental. Chronumental was developed for dealing with very large phylogenies (millions of samples), but lacks some of the functionalities provided by TreeTime (such as estimating uncertainty in date estimates, reconstructing past sequences, re-rooting trees, among others). So, if you are working with less than ~50,000 sequences, we recommend using TreeTime, otherwise Chronumental is a suitable alternative.\nBecause we are dealing with a small number of samples, we will show an example of using TreeTime to scale our tree based on our dates:\ntreetime --tree results/iqtree/uk.treefile --dates sample_annotation.tsv --aln results/mafft/uk_alignment.fa --outdir results/treetime/uk\nAfter running, this tool produces several output files in the specified folder. The main files of interest are:\n\ntimetree.nexus is the new date-scaled tree. NEXUS is another tree file format, which can store more information about the tree compared to the simpler NEWICK format. This format is also supported by FigTree.\ntimetree.pdf is a PDF file with the inferred tree, including a time scale on the x-axis. This can be useful for quick visualisation of the tree.\n\nWe can visualise this tree in FigTree, by opening the file timetree.nexus. The scale of this new tree now corresponds to years (instead of nucleotide substitution rates). We make make several adjustments to this tree, to make it look how we prefer. For example:\n\nLabel the nodes of the tree with the inferred dates: from the left menu click Node Labels and under “Display” select “date”.\nImport the metadata table (sample_annotation.tsv file) and display the results of Nextclade or Pangolin clades/lineages.\nAdjust the scale of the tree to be more intuitive. For example, instead of having the unit of the scale in years, we can change it to months. On the left menu, click Time Scale and change “Scale factor” to 12 (twelve months in a year). Then click Scale Bar and change the “Scale range” to 1. The scale now represents 1 month of time, which may be easier to interpret in this case.\n\nNote that there is uncertainty in the estimates of the internal node dates from TimeTree and these should be interpreted with some caution. The inference of the internal nodes will be better the more samples we have, and across a wider range of times. In our specific case we had samples all from a similar period of time, which makes our dating of internal nodes a little poorer than might be desired.\nMore precise tree dating may be achieved by using public sequences across a range of times or by collecting more samples over time. Time-scaled trees of this sort can therefore be useful to infer if there is a recent spread of a new clade in the population."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#missing-data-problematic-sites",
+ "href": "materials/02-isolates/04-phylogeny.html#missing-data-problematic-sites",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.6 Missing Data & Problematic Sites",
+ "text": "7.6 Missing Data & Problematic Sites\nSo far, we have been using all of our assembled samples in the phylogenetic analysis. However, we know that some of these have poorer quality (for example the “IN01” sample from India had low genome coverage). Although, generally speaking, sequences with missing data are unlikely to substantially affect the phylogenetic results, their placement in the phylogeny will be more uncertain (since several variable sites may be missing data). Therefore, for phylogenetic analysis, it is best if we remove samples with low sequencing coverage, and instead focus on high-quality samples (e.g. with >80% coverage).\nA more serious issue affecting phylogenies is the presence of recurrent errors in certain positions of the genome. One of the regions with a higher prevalence of errors is the start and end of the consensus sequence, which also typically contains many missing data (see example in Figure 2). Therefore, it is common to mask the first and last few bases of the alignment, to avoid including spurious variable sites in the analysis.\n\n\n\n\n\n\nSequence masking\n\n\n\nThe term masking is often used to refer to the process of converting sequence bases to the ambiguous character ‘N’. You may come across this term in the documentation of certain tools, for example: “Positions with less than 20x depth of sequencing are masked.”\nMasks are not limited to depth of sequencing. For example, reference genomes from ENSEMBL are available with masked repeat or low-complexity sequences (e.g. around centromeres, transposon-rich regions, etc.).\nThe term soft masking is also used to refer to cases where, instead of using the ambiguous character ‘N’, sequences are masked with a lowercase. For example:\n>seq_with_soft_masking\nACAGACTGACGCTGTcatgtatgtcgacGATAGGCTGATGGCGAGTGACTCGAG\n>seq_with_hard_masking\nACAGACTGACGCTGTNNNNNNNNNNNNNGATAGGCTGATGGCGAGTGACTCGAG\n\n\nAdditionally, work by Turakhia, de Maio, Thornlow, et al. (2020) has identified several sites that show an unexpected mutation pattern. This includes, for example, mutations that unexpectedly occur multiple times in different parts of the tree (homoplasies) and often coincide with primer binding sites (from amplicon-based protocols) and can even be lab-specific (e.g. due to their protocols and data processing pipelines). The work from this team has led to the creation of a list of problematic sites, which are recommended to be masked before running the phylogenetic analysis.\n\n\n\nExample of errors in phylogenetic inference due to recurrent sequencing errors. Source: Figure 1 in Turakhia, de Maio, Thornlow et al. (2020)\n\n\nSo, let’s try to improve our alignment by masking the problematic sites, which are provided as a VCF file. This file also includes the first and last positions of the genome as targets for masking (positions 1–55 and 29804–29903, relative to the Wuhan-Hu-1 reference genome MN908947.3). The authors also provide a python script for masking a multiple sequence alignment. We have already downloaded these files to our course materials folder, so we can go ahead and use the script to mask our file:\npython scripts/mask_alignment_using_vcf.py --mask -v resources/problematic_sites.vcf -i results/mafft/uk_alignment.fa -o results/mafft/uk_alignment_masked.fa\nIf we open the output file with AliView, we can confirm that the positions specified in the VCF file have now been masked with the missing ‘N’ character.\nWe could then use this masked alignment for our tree-inference, just as we did before.\n\n\n\n\n\n\nUsing Python Scripts\n\n\n\nBioinformaticians often write custom scripts for particular tasks. In this example, the authors of the “problematic sites” wrote a Python script that takes as input the FASTA file we want to mask as well as a VCF with the list of sites to be masked.\nPython scripts are usually run with the python program and often accept options in a similar way to other command-line tools, using the syntax --option (this is not always the case, but most professionally written scripts follow this convention). To see how to use the script we can use the option --help. For our case, we could run:\npython scripts/mask_alignment_using_vcf.py --help"
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#exercises",
+ "href": "materials/02-isolates/04-phylogeny.html#exercises",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.7 Exercises",
+ "text": "7.7 Exercises\n\n\n\n\n\n\nBuilding phylogenies\n\n\n\n\n\n\n\nSo far we have focused our analysis on the samples from the UK. In this exercise you will be able to practice these steps on the samples from India. The steps are similar to what we have done so far, and you can consult the materials in the sections above to go through each exercise.\nIn the following exercises, you can run the commands directly from the command line. But if you feel comfortable with nano, as a bonus, you can try to save the commands in a shell script.\n\nUsing mafft, produce a multiple-sequence alignment from the India consensus sequences in the file data/india_consensus.fa. Save the output in a file named results/mafft/india_alignment.fa.\nUsing iqtree2, infer a phylogenetic tree from this alignment. Save the output with prefix results/iqtree/india.\n\nWhat substitution model did IQ-Tree infer as the most likely for the data?\n\nUsing FigTree, visualise the inferred tree:\n\nOpen the .tree output file.\nImport the annotation table sample_annotation.tsv.\nMake the tip labels display the Nextclade clade instead of the sample names.\nHighlight any clusters of the tree containing WHO variants of concern.\n\nUsing timetree, re-scale the phylogeny using dates (the file sample_annotation.tsv can be used as input to timetree along with the previously-created phylogeny and alignments). Output the result to results/treetime/india\n\nOpen the .nexus output file in FigTree.\nMake the node labels display the date inferred by timetree.\n\nTwo samples in the time-scaled tree appear at the root of the tree: IN05 and IN33. But we would have expected the reference sample (MN908947.3) to be the root of the tree, as it was collected in Dec 2019.\n\nInvestigate what lineages these samples were assigned to.\nGo to https://cov-lineages.org/lineage_list.html and check when those lineages were detected.\nIs the collection date metadata for these samples compatible with the lineage information from the Pangolin website? Can you hypothesise what may have happened with these samples?\n\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nQuestion 1\nWe run our alignment using the following command:\nmafft --6merpair --maxambiguous 0.2 --addfragments data/india_consensus.fa resources/reference/sarscov2.fa > results/mafft/india_alignment.fa\nWe could optionally visualise our alignment using AliView to check that the alignment was successful.\nQuestion 2\nWe can fit a maximum-likelihood tree using the following command:\niqtree2 -s results/mafft/india_alignment.fa --prefix results/iqtree/india\nThis command prints a lot of output on the screen. We can see all this information (and more!) on the output file results/iqtree/india.iqtree, which contains a log of all the analysis done by the program. We could, for example, use the less program to look inside this file. When we do this, we can see the following:\nModelFinder\n-----------\n\nBest-fit model according to BIC: GTR+F+I\nWhich indicates that the substitution model used by IQ-Tree was “GTR+F+I”. This is the same model that was determined for the UK samples, and an explanation of this model is given in the materials above.\nQuestion 3\nUsing FigTree:\n\nGo to File > Open… and browse to the folder with the IQ-Tree output files.\nSelect the file with india.treefile and click Open.\nGo to File > Import annotations… and open the annotation file sample_annotation.tsv.\nOn the menu on the left, click Tip Labels and under “Display” choose the field “nextclade_clade”.\n\nOn this tree, there is a small group of samples classified as “20I (Alpha; V1)”. These samples correspond to the Alpha variant of concern. To highlight these:\n\nChange the “Selection Mode” at the top to “Clade”.\nSelect the branch corresponding to the base of the group of samples classified as Alpha. This should highlight all those branches.\nClick the “Highlight” button at the top and choose a colour.\n\n\nQuestion 4\nThe command to time-scale the tree is:\ntreetime --tree results/iqtree/india.treefile --dates sample_annotation.tsv --aln results/mafft/india_alignment.fa --outdir results/treetime/india\nOnce complete, we can open the india.nexus tree with FigTree. We can annotate the internal nodes of the tree with the dates inferred by treetime by clicking on the Node Labels menu on the left and selecting “Display” to be “date”.\n\nQuestion 5\nTo investigate this strange result, we open the sample_annotation.tsv file in our spreadsheet program. We can see the following information for these two samples:\nname date country year month pango_lineage nextclade_clade\nIN05 2021-01-21 India 2021 1 A.23.1 19B\nIN33 2021-01-21 India 2021 1 A.23.1 19B\nBoth these samples were assigned to Pango lineage “A.23.1”. From the cov-lineages.org website we can see that these were detected as early as 2020-06-08.\nHowever, our samples’ metadata indicate that these samples were collected in January 2021, that is 7 months after these samples were first detected globally.\nThis contractiction explains the strange result in our time-scaled tree. These samples will have mutations that are part of the older lineage “A”, but were annotated to be from 2021. So treetime put them down at the root of the tree.\nThis discrepancy could indicate an issue with the metadata collection, which perhaps is wrong. For example, it could be that these samples were collected months before January 2021, but only sequenced then. And by mistake the date of sequencing was recorded as the date of collection.\nThis is an example how important accurate metadata is for our analysis."
+ },
+ {
+ "objectID": "materials/02-isolates/04-phylogeny.html#summary",
+ "href": "materials/02-isolates/04-phylogeny.html#summary",
+ "title": "7 Building phylogenetic trees",
+ "section": "7.8 Summary",
+ "text": "7.8 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nMethods for phylogenetic inference include parsimony and maximum likelihood. Maximum likelihood methods are preferred because they include more features of the evolutionary process. However, they are computationally more demanding than parsimony-based methods.\nTo build a phylogenetic tree we need a multiple sequence alignment of the sequences we want to infer a tree from.\nIn SARS-CoV-2, alignments are usually done against the Wuhan-Hu-1 reference genome.\nWe can use the software mafft to produce a multiple sequence alignment. The option --addfragments is used to produce an alignment against the reference genome.\nThe software iqtree2 can be used for inferring trees from an alignment using maximum likelihood. This software supports a wide range of substitution models and a method to identify the model that maximizes the likelihood of the data.\nSome of the substituion models that have been used to build global SARS-CoV-2 phylogenies are “GTR+G” and “GTR+I”.\nWe can time-scale trees using sample collection date information. The program treetime can be used to achieve this. For very large sample sizes (>50,000 samples) the much faster program Chronumental can be used instead.\nBefore building a phylogeny, we should be careful to mask problematic sites that can lead to misleading placements of samples in the tree. The SARS-CoV-2 Problematic Sites repository provides with an updated list of sites that should be masked."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#pipeline-overview",
+ "href": "materials/03-case_studies/01-switzerland.html#pipeline-overview",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.1 Pipeline Overview",
+ "text": "8.1 Pipeline Overview\nOur analysis starts with FASTQ files, which will be used with the nf-core/viralrecon Nextflow pipeline. This will give us several quality control metrics essential for our downstream analysis and reporting.\nCritical files output by the pipeline will need to be further processed, including combining our consensus FASTA files and obtaining a list of filtered SNP/indel variants. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of mutations or variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#preparing-files",
+ "href": "materials/03-case_studies/01-switzerland.html#preparing-files",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.2 Preparing Files",
+ "text": "8.2 Preparing Files\nBefore we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. From the data we are starting with, we already have the following directories:\n\ndata → contains the sequencing data in a sub-directory called fast_pass.\nresources → files that were downloaded from public repositories.\nscripts → bash and R scripts used to run the analysis.\n\nWe create two additional directories:\n\nreport → files and documents that we report to our colleagues or upload to public repositories.\nresults → results of the analysis.\n\nYou can create directories from the command line using the mkdir command:\nmkdir results\nmkdir report\n\n8.2.1 Data\nWe start our analysis from FASTQ files generated using the software Guppy v6.1.5 ran in “fast” mode. This software outputs the files to a directory called fastq_pass, with further sub-directories for each sample barcode. This is how it looks like in this case:\nls data/fastq_pass\nbarcode01 barcode08 barcode15 barcode22 barcode29 barcode36 barcode43 barcode50 barcode57\nbarcode02 barcode09 barcode16 barcode23 barcode30 barcode37 barcode44 barcode51 barcode58\nbarcode03 barcode10 barcode17 barcode24 barcode31 barcode38 barcode45 barcode52 barcode59\nbarcode04 barcode11 barcode18 barcode25 barcode32 barcode39 barcode46 barcode53 barcode60\nbarcode05 barcode12 barcode19 barcode26 barcode33 barcode40 barcode47 barcode54 barcode61\nbarcode06 barcode13 barcode20 barcode27 barcode34 barcode41 barcode48 barcode55 barcode62\nbarcode07 barcode14 barcode21 barcode28 barcode35 barcode42 barcode49 barcode56 barcode63\n\n\n8.2.2 Metadata\nMetadata for these samples is available in the file sample_info.csv. Here is some of the information we have available for these samples:\n\nsample → the sample ID.\ncollection_date → the date of collection for the sample in the format YYYY-MM-DD.\ncountry → the country of origin for this sample.\nlatitude/longitude → coordinates for sample location (optional).\nsequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).\nsequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).\namplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)\nSpecific columns for Oxford Nanopore data, which are essential for the bioinformatic analysis:\n\nont_pore → the version of the pores.\nont_guppy_version → the version of the Guppy software used for basecalling.\nont_guppy_mode → the basecalling mode used with Guppy."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#consensus-assembly",
+ "href": "materials/03-case_studies/01-switzerland.html#consensus-assembly",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.3 Consensus Assembly",
+ "text": "8.3 Consensus Assembly\n\n\n\n\n\n\nNote\n\n\n\nSee Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.\n\n\nThe first step in the bioinformatic analysis is to run the nf-core/viralrecon pipeline.\n\n8.3.1 Samplesheet\nBut first we need to prepare our input files. For Nanopore data, we need a samplesheet CSV file with two columns, indicating sample name (first column) and the respective barcode number (second column).\nWe produced this table in Excel and saved it as a CSV file. Here are the top few rows of the file:\nhead samplesheet.csv\nsample,barcode\nCH01,1\nCH02,2\nCH03,3\nCH04,4\nCH05,5\nCH06,6\nCH07,7\nCH08,8\nCH09,9\nCH10,10\n\n\n8.3.2 Running Viralrecon\nNow we are ready to run the nf-core/viralrecon pipeline (see Section 4.3 for details). We saved our command in a script (scripts/01-run_viralrecon.sh), which we created with the command line text editor nano. This ensures that our analysis is reproducible and traceable (we can go back to the script to see how the analysis was run).\nFirst, we activate our software environment, to ensure Nextflow is available to us:\nmamba activate nextflow\nThen, we run our script with:\nbash scripts/01-run_viralrecon.sh\nWhich will start executing the pipeline.\nFor reference, here is the command included in that script:\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '16.GB' --max_cpus 8 \\\n --platform nanopore \\\n --input samplesheet.csv \\\n --fastq_dir data/fastq_pass/ \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --artic_minion_caller medaka \\\n --artic_minion_medaka_model r941_min_fast_g303 \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\nIn this case, we used the medaka model r941_min_fast_g303, because that is the latest one available (even though our Guppy version is 6.1.5). We also restricted our --max_memory and --max_cpus due to the size of our processing computers. If a larger computer was available, we could have used higher values for these parameters.\n\n\n\n\n\n\nThe \\ in long commands\n\n\n\nIn the command above, you will notice several \\ at the end of each line. This is indicating that we want to continue writing our command in the next line. Notice that the last line does not include \\, because that is the end of the command. This is very useful when commands are very long, because it makes the code more readable."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#consensus-quality",
+ "href": "materials/03-case_studies/01-switzerland.html#consensus-quality",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.4 Consensus Quality",
+ "text": "8.4 Consensus Quality\n\n\n\n\n\n\nNote\n\n\n\nSee Section 5.2, if you need to revise how to assess the quality of consensus sequences.\n\n\n\n8.4.1 General Metrics\nWe used the MultiqQC report to assess the initial quality of our samples. The quality report can be found in results/viralrecon/multiqc/medaka/multiqc_report.html.\nWe paid particular attention to:\n\nNumber of reads mapped to the reference genome.\nMedian depth of coverage.\nPercentage of the genome with missing bases (‘N’).\nNumber of SNP + Indel variants.\n\n\n\n\nExample MultiQC output, highlighting the main columns with quality information that will be collected for the final report.\n\n\nWe noted that:\n\n9 samples had more than 15% missing bases.\nAll samples had median depth of coverage greater than 20x.\nThere was some systematic dropout for some amplicons, in particular nCoV-2019_64 had very low amplification in several of the samples. Of note was also nCoV-2019_73, and other neighbouring amplicons.\n\n\n\n\nAmplicon depth of sequencing (or coverage) across samples, from the MultiQC report. Darker regions indicate systematic amplicon dropout, probably due to issues during the PCR amplification.\n\n\nBesides the MultiQC report, the pipeline also outputs a CSV file with collected summary metrics (equivalent to the first table on the report): results/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv. We will use this file later to join this information with our metadata and lineage assignment using the R software (detailed in “Integration & Visualisation” section, below).\n\n\n8.4.2 Variants\nWe also looked at the table of variants obtained from the pipeline. This is output in results/viralrecon/medaka/variants_long_table.csv. This table can be very useful to keep track of particular mutations that may be increasing over time. Later, we will tidy this table to attach to our reported results (“Integration & Visualisation” section).\nBut for now, we will explore this table to address a few more quality-related questions. We opened this table in Excel to answer the following:\n\nWhere there samples with a high number of intermediate allele frequencies? This could indicate mixed samples due to cross-contamination.\nWhere there samples with frameshift mutations? These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.\n\nAll the variants in our samples had an alternative allele frequency (AF column) greater than 68%, which is a good indication that there were no mixed samples. We did note two samples – CH13 and CH49 – had a frameshift mutation each, in the Spike and ORF1a proteins, respectively. This may be due to a sequencing error, as it is not observed in any other samples. The inclusion of this error may not affect our downstream analysis significantly, but we make a note of these two samples, to check if their lineage assignment and phylogenetic analysis makes sense later on (or whether they appear as outliers, indicating broader sequencing quality issues).\n\n\n\nVariants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases.\n\n\n\n\n8.4.3 Clean FASTA\nThe pipeline outputs the consensus sequences in results/viralrecon/medaka/*.consensus.fasta (one file for each sample). For downstream analysis, it is convenient to combine all these sequences into a single file, and also clean the sequence names (to remove some text – “/ARTIC/medaka MN908947.3” –, which is added by the medaka variant caller).\nWe created a new script to clean our consensus FASTA files, which we ran with bash scripts/02-clean_fasta.sh:\n#!/bin/bash \n\n# combine and clean FASTA files\ncat results/viralrecon/medaka/*.consensus.fasta | sed 's|/ARTIC/medaka MN908947.3||' > report/consensus.fa\nThis command does two things:\n\nCombine all our FASTA consensus sequences into a single file (using cat).\nClean the sequence names (using sed).\n\nThe output was saved as a new FASTA file: report/consensus.fa.\n\n\n8.4.4 Missing Intervals\nAs a further quality check, we also generated a table of missing intervals (indicated by the N character in the FASTA sequences). We used the seqkit software to achieve this.\nFirst, we activate our software environment:\nmamba activate seqkit\nThen, we ran the script bash scripts/03-missing_intervals.sh, which includes the following command:\nseqkit locate -i -P -G -M -r -p \"N+\" report/consensus.fa > results/missing_intervals.tsv\nThis software outputs a tab-delimited table, which we saved as results/missing_intervals.tsv. The table looks like this (only the top few rows are shown):\nseqID patternName pattern strand start end\nCH01 N+ N+ + 1 54\nCH01 N+ N+ + 1193 1264\nCH01 N+ N+ + 4143 4322\nCH01 N+ N+ + 6248 6294\nCH01 N+ N+ + 7561 7561\nCH01 N+ N+ + 9243 9311\nCH01 N+ N+ + 10367 10367\nCH01 N+ N+ + 11361 11370\nCH01 N+ N+ + 13599 13613\nWe opened this file missing_intervals.tsv in Excel and quickly calculated the length of each interval. We noted that two samples – CH07 and CH59 – both have a continuous interval of 5130 missing bases between positions 19580 and 24709. This includes part of the ORF1a gene and nearly all of the S (Spike) gene. This may be due to a poor amplification of one of the PCR amplicons and may affect the interpretation of the results for these two samples. We make a note of these samples as being possibly problematic in downstream analysis steps."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#downstream-analyses",
+ "href": "materials/03-case_studies/01-switzerland.html#downstream-analyses",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.5 Downstream Analyses",
+ "text": "8.5 Downstream Analyses\nBased on the clean consensus sequences, we then perform several downstream analysis.\n\n8.5.1 Lineage Assignment\n\n\n\n\n\n\nNote\n\n\n\nSee Section 6.1, if you need to revise how lineage assignment works.\n\n\nAlthough the Viralrecon pipeline runs Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). An up-to-date run of both of these tools can be done using each of their web applications:\n\nclades.nextstrain.org\npangolin.cog-uk.io\n\nHowever, for automation, reproducibility and traceability purposes, we used the command line versions of these tools, and included their analysis in two scripts.\n\nNextcladePangolin\n\n\nFor Nextclade, we first activate the software environment:\nmamba activate nextclade\nAnd then we ran the script bash scripts/04-nextclade.sh, which contains the following commands:\n# get nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa\nThe first command downloads the latest version of the Nextclade background data using nextclade dataset. We use that data as input to the second command (nextclade run) to make sure it runs with the most up-to-date lineages.\n\n\nFor Pangolin, we first activate the software environment:\nmamba activate pangolin\nAnd then we ran the script bash/04-pangolin.sh, which contains the following commands:\n# update pangolin data\npangolin --update-data\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa\nSimilarly to before, we first ran pangolin --update-data to ensure we were using the latest lineages available. We can check the version of the data used with pangolin --all-versions (at the time we ran this we had pangolin-data: 1.23.1).\n\n\n\nBoth of these tools output CSV files, which can be open in Excel for further examination.\nOpening the pangolin results (results/pangolin/pango_report.csv), we noticed that two samples – CH07 and CH59 – failed the QC due to high fraction of missing data. These are the same two samples that had a large gap of missing data in the previous section. Several other samples were classified as “Probable Omicron”, which from the “scorpio_notes” column we can see may be because too many of the expected mutations had missing (ambiguous) bases in those consensus sequences.\nOpening the nextclade results (results/nextclade/nextclade.tsv), we noticed that 23 samples were assigned a QC status of “bad”, mostly due to high percentage of missing data (nextclade uses a stringent threshold of 3000 sites, or ~10%, missing data).\nLike before, we will do further analysis (and visualisation) of these data using the software R, in the section “Integration & Visualisation”, detailed below.\n\n\n8.5.2 Phylogeny\n\n\n\n\n\n\nNote\n\n\n\nSee Section 7.1, if you need to revise how to build phylogenetic trees.\n\n\nAlthough a tool such as Nextclade can place our samples in a global phylogeny context, sometimes it may be convient to build our own phylogenies. This requires three steps:\n\nProducing a multiple sequence alignment from all consensus sequences.\nTree inference.\nTree visualisation and annotation.\n\nBefore our analysis, we first activated our software environment:\nmamba activate phylo\nWe performed the first two steps with the following script, which we ran with bash scripts/05-phylogeny.sh:\n# alignment\nmkdir -p results/mafft\nmafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa\n\n# tree inference\nmkdir -p results/iqtree\niqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensus\nThe output of iqtree includes a tree file, which can be visualised using FigTree (or online using Microreact). The figure below shows an example of an annotated tree, where we highlight the main VOCs detected. This annotation was done based on the file that we generate in the next section (“Integration & Visualisation”), so those steps would have to be done first.\nWe can see that our samples fall broadly into two large clusters, which correlate with the VOC classification for these samples.\n\n\n\nPhylogenetic tree for our samples. The top cluster (purple) highlights samples classified as Omicron (BA.1-related) and the bottom cluster (pink) samples classified as Delta (AY lineage, also known as B.1.617.2). The tips of the tree are coloured according to Nextclade’s QC status: green = “good”; blue = “mediocre”; red = “bad”.\n\n\n\n\n8.5.3 Clustering\nWe identified groups of similar sequences in our data using the software civet (Cluster Investigation and Virus Epidemiology Tool). This software compares our samples with a background dataset of our choice, which givus us more context for our analysis. In this case we are using the example background data that comes with civet. However, in a real-world analysis, it would have been ideal to choose local samples as background data. For example, we could download samples from Switzerland from around the time period of our sample collection, from GISAID following the instructions on the civet documentation (you need an account on GISAID to obtain these data).\nFor this example, we already prepared civet background dataset saved in resources/civet_background_data.\nBefore our analysis, we first activate our software environment:\nmamba activate civet\nThen, we ran the script bash scripts/06-civet.sh, which contains the following code:\n# run civet analysis\ncivet \\\n -i sample_info.csv \\\n -f report/consensus.fa \\\n -icol sample \\\n -idate collection_date \\\n -d resources/civet_background_data/ \\\n -o results/civet\nThe result of this analysis includes an interactive HTML report (in results/civet/civet.html). We can see that our samples were grouped into 2 catchments, using this background data. This makes sense from our previous lineage/variant analysis: the two catchments correlate with the two main variants in these data (Omicron and Delta).\n\n\n\nExample of results from civet report for catchment 2, showing the phylogeny in the context of the samples in the background data (coloured according to their lineages).\n\n\nCivet also outputs a CSV file (results/civet/master_metadata.csv), which includes the catchment that each sample was assigned to. We will use this CSV file later to integrate this information with other parts of our analysis, in R, detailed in the “Integration & Visualisation” section."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#integration-visualisation",
+ "href": "materials/03-case_studies/01-switzerland.html#integration-visualisation",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.6 Integration & Visualisation",
+ "text": "8.6 Integration & Visualisation\nAt this point in our analysis, we have several tables with different pieces of information:\n\nsample_info.csv → the original table with metadata for our samples.\nresults/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.\nresults/nextclade/nextclade.tsv → the results from Nextclade.\nresults/pangolin/pango_report.csv → the results from Pangolin.\nresults/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.\n\nTo consolidate our analysis, we tidied and integrated the information from across these different files, into a single table using the software R. The script used to do this is in scripts/07-data_integration.R. Because this is an R script, we opened it in RStudio to execute the code.\nThe output of our script is a new tab-delimited table, which we saved in report/consensus_metrics.tsv, and contains the following columns:\n\nsample → sample ID.\ncollection_date → date of collection day.\ncollection_week → date of collection week (useful for summarising/visualising counts per-week).\ncountry → country of origin.\nlatitude/longitude → latitude and longitude of collection.\nn_mapped_reads → number of mapped reads.\nmedian_depth → median depth.\npct_missing → percentage of missing data.\npct_coverage → percentage of coverage.\nn_variants → number of SNP + indel variants detected.\nnextclade → nextclade clade.\nqc_status → QC status as determined by Nextclade (“bad”, “mediocre”, “good”).\nlineage → Pangolin lineage.\nwho_variant → variant of concern designation.\ncatchment → catchment group from Civet.\n\nThis table, which aggregates information from many of the tools we used, was then used to produce different visualisations of our analysis. These visualisations were also done using the R software (scripts/08-visualisation.R), and integrated into a report, shown below."
+ },
+ {
+ "objectID": "materials/03-case_studies/01-switzerland.html#bonus-full-workflow",
+ "href": "materials/03-case_studies/01-switzerland.html#bonus-full-workflow",
+ "title": "8 Switzerland (Nanopore)",
+ "section": "8.7 Bonus: Full Workflow",
+ "text": "8.7 Bonus: Full Workflow\nAlthough we have ran each of the steps of our analysis individually (each in their own script), now that we have everything working, we could integrate all these steps into a single “master” script:\n#!/bin/bash\n\n# make mamba activate command available\neval \"$(conda shell.bash hook)\"\nsource $(mamba info --base)/etc/profile.d/mamba.sh\n\n# make report directory\nmkdir -p report\n\nmamba activate nextflow\n\n# run viralrecon\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '16.GB' --max_cpus 8 \\\n --platform nanopore \\\n --input samplesheet.csv \\\n --fastq_dir data/fastq_pass/ \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --artic_minion_caller medaka \\\n --artic_minion_medaka_model r941_min_fast_g303 \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\n\n# combine and clean FASTA files\ncat results/viralrecon/medaka/*.consensus.fasta | sed 's|/ARTIC/medaka MN908947.3||' > report/consensus.fa\n\nmamba activate seqkit\n\n# create missing bases TSV file\nseqkit locate -i -P -G -M -r -p \"N+\" report/consensus.fa > results/missing_intervals.tsv\n\nmamba activate nextclade\n\n# get nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa\n\nmamba activate pangolin\n\n# update pangolin data\npangolin --update-data\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa\n\nmamba activate phylo\n\n# alignment\nmkdir -p results/mafft\nmafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa\n\n# tree inference\nmkdir -p results/iqtree\niqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensus\n\n# data integration and cleaning\nRscript scripts/07-data_integration.R\nNotice that we included the R script that does the data cleaning here, using the Rscript program that allows to execute an R script from the command-line.\nHaving this “master” script, we could run all these steps from start-to-finish with a single command, which can be very useful if you want to fully automate your analysis across multiple runs."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#pipeline-overview",
+ "href": "materials/03-case_studies/02-southafrica.html#pipeline-overview",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.1 Pipeline Overview",
+ "text": "9.1 Pipeline Overview\nOur analysis starts with FASTQ files, which will be used with the nf-core/viralrecon Nextflow pipeline. This will give us several quality control metrics essential for our downstream analysis and reporting.\nCritical files output by the pipeline will need to be further processed, including combining our consensus FASTA files and obtaining a list of filtered SNP/indel variants. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of mutations or variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#preparing-files",
+ "href": "materials/03-case_studies/02-southafrica.html#preparing-files",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.2 Preparing Files",
+ "text": "9.2 Preparing Files\nBefore we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. From the data we are starting with, we already have the following directories:\n\ndata → contains the sequencing data in a sub-directory called reads.\nresources → files that were downloaded from public repositories.\nscripts → bash and R scripts used to run the analysis.\n\nWe create two additional directories:\n\nreport → files and documents that we report to our colleagues or upload to public repositories.\nresults → results of the analysis.\n\nYou can create directories from the command line using the mkdir command:\nmkdir results\nmkdir report\n\n9.2.1 Data\nWe start our analysis from FASTQ files generated by the Illumina sequencer. As this is paired-end sequencing, we have two files per sample (with suffix _1 and _2):\nls data/reads\nSRR17051908_1.fastq.gz SRR17051953_1.fastq.gz SRR17461700_1.fastq.gz SRR17712594_1.fastq.gz\nSRR17051908_2.fastq.gz SRR17051953_2.fastq.gz SRR17461700_2.fastq.gz SRR17712594_2.fastq.gz\nSRR17051916_1.fastq.gz SRR17054503_1.fastq.gz SRR17461712_1.fastq.gz SRR17712607_1.fastq.gz\nSRR17051916_2.fastq.gz SRR17054503_2.fastq.gz SRR17461712_2.fastq.gz SRR17712607_2.fastq.gz\nSRR17051923_1.fastq.gz SRR17088917_1.fastq.gz SRR17701832_1.fastq.gz SRR17712711_1.fastq.gz\nSRR17051923_2.fastq.gz SRR17088917_2.fastq.gz SRR17701832_2.fastq.gz SRR17712711_2.fastq.gz\nSRR17051932_1.fastq.gz SRR17088924_1.fastq.gz SRR17701841_1.fastq.gz SRR17712779_1.fastq.gz\nSRR17051932_2.fastq.gz SRR17088924_2.fastq.gz SRR17701841_2.fastq.gz SRR17712779_2.fastq.gz\nSRR17051935_1.fastq.gz SRR17088928_1.fastq.gz SRR17701890_1.fastq.gz SRR17712994_1.fastq.gz\nSRR17051935_2.fastq.gz SRR17088928_2.fastq.gz SRR17701890_2.fastq.gz SRR17712994_2.fastq.gz\nSRR17051951_1.fastq.gz SRR17088930_1.fastq.gz SRR17712442_1.fastq.gz SRR17712997_1.fastq.gz\nSRR17051951_2.fastq.gz SRR17088930_2.fastq.gz SRR17712442_2.fastq.gz SRR17712997_2.fastq.gz\n\n\n9.2.2 Metadata\nMetadata for these samples is available in the file sample_info.csv. Here is some of the information we have available for these samples:\n\nsample → the sample ID.\ncollection_date → the date of collection for the sample in the format YYYY-MM-DD.\ncountry → the country of origin for this sample.\ngeo_loc_region → the region within the country where the sample was collected.\nlatitude/longitude → coordinates for sample location (in this case we’re only given a single coordinate for the whole country - in a real setting you may want to collect a precise location).\nsequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).\nsequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).\namplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)"
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#consensus-assembly",
+ "href": "materials/03-case_studies/02-southafrica.html#consensus-assembly",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.3 Consensus Assembly",
+ "text": "9.3 Consensus Assembly\n\n\n\n\n\n\nNote\n\n\n\nSee Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.\n\n\nThe first step in the bioinformatic analysis is to run the nf-core/viralrecon pipeline. But first we need to prepare our input files.\n\n9.3.1 Samplesheet\nFor Illumina data, we need a samplesheet CSV file with three columns, indicating sample name (first column) and the respective FASTQ file paths for read 1 (second column) and read 2 (third column).\nBecause our FASTQ file names are not very user-friendly, we used some command-line tricks to help us produce this table:\n# list read 1 files and save output in a temporary file\nls data/reads/*_1.fastq.gz > read1_filenames.txt\n\n# list read 2 files and save output in a temporary file\nls data/reads/*_2.fastq.gz > read2_filenames.txt\n\n# initiate a file with column names\necho \"fastq_1,fastq_2\" > samplesheet.csv\n\n# paste the two temporary files together, using comma as a delimiter\npaste -d \",\" read1_filenames.txt read2_filenames.txt >> samplesheet.csv\n\n# remove the two temporary files\nrm read1_filenames.txt read2_filenames.txt\nThese commands resulted in creating a file called samplesheet.csv, which contains the following:\nhead samplesheet.csv\nfastq_1,fastq_2\ndata/reads/SRR17051908_1.fastq.gz,data/reads/SRR17051908_2.fastq.gz\ndata/reads/SRR17051916_1.fastq.gz,data/reads/SRR17051916_2.fastq.gz\ndata/reads/SRR17051923_1.fastq.gz,data/reads/SRR17051923_2.fastq.gz\ndata/reads/SRR17051932_1.fastq.gz,data/reads/SRR17051932_2.fastq.gz\ndata/reads/SRR17051935_1.fastq.gz,data/reads/SRR17051935_2.fastq.gz\ndata/reads/SRR17051951_1.fastq.gz,data/reads/SRR17051951_2.fastq.gz\ndata/reads/SRR17051953_1.fastq.gz,data/reads/SRR17051953_2.fastq.gz\ndata/reads/SRR17054503_1.fastq.gz,data/reads/SRR17054503_2.fastq.gz\ndata/reads/SRR17088917_1.fastq.gz,data/reads/SRR17088917_2.fastq.gz\nSo, we programmatically created the last two columns of our file. We then opened this CSV file in Excel to add another column “sample” where we included our sample names and saved the file again as a CSV format. Here are the top few rows of the final file:\nhead samplesheet.csv\nsample,fastq_1,fastq_2\nZA01,data/reads/SRR17051908_1.fastq.gz,data/reads/SRR17051908_2.fastq.gz\nZA02,data/reads/SRR17051923_1.fastq.gz,data/reads/SRR17051923_2.fastq.gz\nZA03,data/reads/SRR17051916_1.fastq.gz,data/reads/SRR17051916_2.fastq.gz\nZA04,data/reads/SRR17051953_1.fastq.gz,data/reads/SRR17051953_2.fastq.gz\nZA05,data/reads/SRR17051951_1.fastq.gz,data/reads/SRR17051951_2.fastq.gz\nZA06,data/reads/SRR17051935_1.fastq.gz,data/reads/SRR17051935_2.fastq.gz\nZA07,data/reads/SRR17051932_1.fastq.gz,data/reads/SRR17051932_2.fastq.gz\nZA08,data/reads/SRR17054503_1.fastq.gz,data/reads/SRR17054503_2.fastq.gz\nZA09,data/reads/SRR17088930_1.fastq.gz,data/reads/SRR17088930_2.fastq.gz\n\n\n9.3.2 Running Viralrecon\nNow we are ready to run the nf-core/viralrecon pipeline (see Section 4.3 for details). We saved our command in a script (scripts/01-run_viralrecon.sh), which we created with the command line text editor nano. This ensures that our analysis is reproducible and traceable (we can go back to the script to see how the analysis was run).\nFirst, we activate our software environment, to ensure Nextflow is available to us:\nmamba activate nextflow\nThen, we run our script with:\nbash scripts/01-run_viralrecon.sh\nWhich will start executing the pipeline.\nFor reference, here is the command included in that script:\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '15.GB' --max_cpus 8 \\\n --platform illumina \\\n --input samplesheet.csv \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\n\n\n\n\n\n\nThe \\ in long commands\n\n\n\nIn the command above, you will notice several \\ at the end of each line. This is indicating that we want to continue writing our command in the next line. Notice that the last line does not include \\, because that is the end of the command. This is very useful when commands are very long, because it makes the code more readable.\n\n\nAfter running the pipeline, we notice the following message:\n-[nf-core/viralrecon] 2 samples skipped since they failed Bowtie2 1000 mapped read threshold:\n 201: ZA09\n 176: ZA12\nThis indicates that two samples - ZA09 and ZA12 - had very few reads aligned to the SARS-CoV-2 genome. Could the reason be that they had very few reads to start with? We can quickly investigate this hypothesis by counting the number of lines in the FASTQ files from these samples:\n# FASTQ file for ZA09\nzcat data/reads/SRR17088930_1.fastq.gz | wc -l\n698044\nSince each sequence takes 4 lines in a FASTQ format, this indicates that this sample had 698044/4 = 124511 reads. That’s certainly more than 1000 reads, so the reason the sample failed must be something else. We will investigate this further in the next section."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#consensus-quality",
+ "href": "materials/03-case_studies/02-southafrica.html#consensus-quality",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.4 Consensus Quality",
+ "text": "9.4 Consensus Quality\n\n\n\n\n\n\nNote\n\n\n\nSee Section 5.2, if you need to revise how to assess the quality of consensus sequences.\n\n\n\n9.4.1 General Metrics\nWe used the MultiqQC report to assess the initial quality of our samples. The quality report can be found in results/viralrecon/multiqc/multiqc_report.html.\nWe paid particular attention to:\n\nNumber of reads mapped to the reference genome.\nMedian depth of coverage.\nPercentage of the genome with missing bases (‘N’).\nNumber of SNP + Indel variants.\n\n\n\n\nExample MultiQC output, highlighting the main columns with quality information that will be collected for the final report. Note that some columns have been ommited here for clarity.\n\n\nWe noted that:\n\n2 samples - ZA09 and ZA12 - completely failed. This was already noted after we ran the pipeline, above. The reason seems to be because these samples have a very low percentage of non-human reads (less than 2%), indicating that the sequenced material was mostly human. Because of this, there were not enough reads to assemble a genome.\n2 other samples - ZA10 and ZA23 - had > 90% missing bases, also indicating a very poor assembly. The reason was the same as above, both samples had low % of non-human reads.\n1 samples - ZA14 - had ~39% missing bases. In this case the reason was not a low number of mapped reads. For example, sample ZA13 had a very similar median depth of coverage (121 vs 161 in ZA14) and even fewer mapped reads (24k vs 60k in ZA14). But ZA13 only had 2% missing bases. However, upon inspection of the amplicon heatmap, we detected that several amplicons were not properly amplified in ZA14 compared to ZA13. Therefore, the reason for high % missing bases in ZA14 was due to low amplification efficiency in this sample.\nThere was some systematic dropout for some amplicons, in particular nCoV-2019_64 had very low amplification in several of the samples. Of note was also nCoV-2019_73, and other neighbouring amplicons.\n\n\n\n\nAmplicon depth of sequencing (or coverage) across samples, from the MultiQC report. Darker regions indicate systematic amplicon dropout, probably due to issues during the PCR amplification.\n\n\nBesides the MultiQC report, the pipeline also outputs a CSV file with collected summary metrics (equivalent to the first table on the report): results/viralrecon/multiqc/summary_variants_metrics_mqc.csv. We will use this file later to join this information with our metadata and lineage assignment using the R software (detailed in “Integration & Visualisation” section, below).\n\n\n9.4.2 Variants\nWe also looked at the table of variants obtained from the pipeline. This is output in results/viralrecon/variants/ivar/variants_long_table.csv. This table can be very useful to keep track of particular mutations that may be increasing over time. Later, we will tidy this table to attach to our reported results (“Integration & Visualisation” section).\nBut for now, we will explore this table to address a few more quality-related questions. We opened this table in Excel to answer the following:\n\nWhere there samples with a high number of intermediate allele frequencies? This could indicate mixed samples due to cross-contamination.\n\nWhere there samples with frameshift mutations? These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.\n\nBy manual inspection of this table (and using the “filter” feature in Excel), we found 74 variants with alternative allele frequency less than 75%. These were spread across samples, all samples having less than 10 such low-frequency mutations, and most samples having less than 5. Sample ZA23 - which we had previously highlighted has having a high % missing bases (39%) - had 8 low-frequency mutations out of a total of 48 mutations in this sample (~16%), suggesting further quality issues in this sample.\n\n\n\nVariants table output by viralrecon, with a summary of the main columns of interest. Note that this table also includes a column with lineage assignment (not shown in this snapshot). Remember that at this stage we mostly ignore this column, as viralrecon does not use the most up-to-date version of the lineage databases."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#clean-fasta",
+ "href": "materials/03-case_studies/02-southafrica.html#clean-fasta",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.5 Clean FASTA",
+ "text": "9.5 Clean FASTA\nThe pipeline outputs the consensus sequences in results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa (one file for each sample). For downstream analysis, it is convenient to combine all these sequences into a single file, and also clean the sequence names (to remove some text – ” MN908947.3” –, which is added by the bcftools variant caller).\nWe created a new script to clean our consensus FASTA files, which we ran with bash scripts/02-clean_fasta.sh:\n\n# combine and clean FASTA files\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa | sed 's| MN908947.3||' > report/consensus.fa\nThis command does two things:\n\nCombine all our FASTA consensus sequences into a single file (using cat).\nClean the sequence names (using sed).\n\nThe output was saved as a new FASTA file: report/consensus.fa.\n\n\n9.5.1 Missing Intervals\nAs a further quality check, we also generated a table of missing intervals (indicated by the N character in the FASTA sequences). We used the seqkit software to achieve this.\nFirst, we activate our software environment:\nmamba activate seqkit\nThen, we ran the script bash scripts/03-missing_intervals.sh, which includes the following command:\nseqkit locate -i -P -G -M -r -p \"N+\" report/consensus.fa > results/missing_intervals.tsv\nThis software outputs a tab-delimited table, which we saved as results/missing_intervals.tsv. The table looks like this (only the top few rows are shown):\nseqID patternName pattern strand start end\nZA01 N+ N+ + 1 54\nZA01 N+ N+ + 22771 22926\nZA01 N+ N+ + 23603 23835\nZA01 N+ N+ + 26948 26948\nZA01 N+ N+ + 26968 27137\nZA01 N+ N+ + 29801 29867\nZA02 N+ N+ + 1 54\nZA02 N+ N+ + 22771 22921\nZA02 N+ N+ + 23603 23835\nWe opened this file missing_intervals.tsv in Excel and quickly calculated the length of each interval. We noted that two samples - ZA10 and ZA23 - both have a continuous interval of over 18kb missing bases, which is not surprising as we had already identified these samples has having >90% missing data. We make a note of these samples as being possibly problematic in downstream analysis steps."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#downstream-analyses",
+ "href": "materials/03-case_studies/02-southafrica.html#downstream-analyses",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.6 Downstream Analyses",
+ "text": "9.6 Downstream Analyses\nBased on the clean consensus sequences, we then perform several downstream analysis.\n\n9.6.1 Lineage Assignment\n\n\n\n\n\n\nNote\n\n\n\nSee Section 6.1, if you need to revise how lineage assignment works.\n\n\nAlthough the Viralrecon pipeline runs Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). An up-to-date run of both of these tools can be done using each of their web applications:\n\nclades.nextstrain.org\npangolin.cog-uk.io\n\nHowever, for automation, reproducibility and traceability purposes, we used the command line versions of these tools, and included their analysis in two scripts.\n\nNextcladePangolin\n\n\nFor Nextclade, we first activate the software environment:\nmamba activate nextclade\nAnd then we ran the script bash scripts/04-nextclade.sh, which contains the following commands:\n# get nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa\nThe first command downloads the latest version of the Nextclade background data using nextclade dataset. We use that data as input to the second command (nextclade run) to make sure it runs with the most up-to-date lineages.\n\n\nFor Pangolin, we first activate the software environment:\nmamba activate pangolin\nAnd then we ran the script bash/04-pangolin.sh, which contains the following commands:\n# update pangolin data\npangolin --update-data\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa\nSimilarly to before, we first ran pangolin --update-data to ensure we were using the latest lineages available. We can check the version of the data used with pangolin --all-versions (at the time we ran this we had pangolin-data: 1.23.1).\n\n\n\nBoth of these tools output CSV files, which can be open in Excel for further examination.\nOpening the pangolin results (results/pangolin/pango_report.csv), we noticed that the two problematic samples – ZA10 and ZA23 – failed the QC due to high fraction of missing data. The other samples all seemed to have been assigned to “Omicron” variant with a high support.\nOpening the nextclade results (results/nextclade/nextclade.tsv), we noticed that the two problematic samples were classified as “recombinant”! We know from our quality control that we should not trust this assessment, and that most likely these are bad quality samples, not true recombinant lineages. Nextclade is more relaxed in assigning samples to lineages, so we should always check the QC status as well. We will notice that both of these samples were assigned QC status “bad”, due to their high percentage of missing data (nextclade uses a stringent threshold of 3000 sites, or ~10%, missing data). Two other samples had “bad” QC status. Sample ZA14 due to high % of missing data, and sample ZA18 from a mixture of a high number of private mutations and the presence of a frameshift mutation in ORF1b.\nLike before, we will do further analysis (and visualisation) of these data using the software R, in the section “Integration & Visualisation”, detailed below.\n\n\n9.6.2 Phylogeny\n\n\n\n\n\n\nNote\n\n\n\nSee Section 7.1, if you need to revise how to build phylogenetic trees.\n\n\nAlthough a tool such as Nextclade can place our samples in a global phylogeny context, sometimes it may be convient to build our own phylogenies. This requires three steps:\n\nProducing a multiple sequence alignment from all consensus sequences.\nTree inference.\nTree visualisation and annotation.\n\nBefore our analysis, we first activated our software environment:\nmamba activate phylo\nWe performed the first two steps with the following script, which we ran with bash scripts/05-phylogeny.sh:\n# alignment\nmkdir -p results/mafft\nmafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa\n\n# tree inference\nmkdir -p results/iqtree\niqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensus\nThe output of iqtree includes a tree file, which can be visualised using FigTree (or online using Microreact). The figure below shows our tree, which shows all our samples fall mostly clustering together. This makes sense, as all our samples were classified as “Omicron (BA.1-related)”.\nIt is worth noting that the samples ZA10, ZA14 and ZA23 are not included in this phylogeny, as they contained >20% missing data (we used that threshold with MAFFT alignment, option --maxambiguous 0.2). Also, note that sample ZA18, which Nextclade had identified as “bad” QC (due to excess private mutations) also appears slightly separated from the other samples in the tree (the sample in red on the tree). The sample still clusters well with the rest, suggesting we can probably trust that it is indeed an Omicron variant, however the excess of private mutations (which may be due to sequencing errors) is making it stand apart from the others in the phylogeny.\n\n\n\nPhylogenetic tree for our samples. All samples were classified as lineage BA.1 (and some of its sub-lineages), corresponding to the Omicron VOC. The tips of the tree are coloured according to Nextclade’s QC status: green = “good”; blue = “mediocre”; red = “bad”.\n\n\n\n\n9.6.3 Clustering\nWe identified groups of similar sequences in our data using the software civet (Cluster Investigation and Virus Epidemiology Tool). This software compares our samples with a background dataset of our choice, which givus us more context for our analysis. In this case we are using the example background data that comes with civet. However, in a real-world analysis, it would have been ideal to choose local samples as background data. For example, we could download samples from South Africa from around the time period of our sample collection, from GISAID following the instructions on the civet documentation (you need an account on GISAID to obtain these data).\nFor this example, we already prepared civet background dataset saved in resources/civet_background_data.\nBefore our analysis, we first activate our software environment:\nmamba activate civet\nThen, we ran the script bash scripts/06-civet.sh, which contains the following code:\n# run civet analysis\ncivet \\\n -i sample_info.csv \\\n -f report/consensus.fa \\\n -icol sample \\\n -idate collection_date \\\n -d resources/civet_background_data/ \\\n -o results/civet\nThe result of this analysis includes an interactive HTML report (in results/civet/civet.html). We can see that our samples were grouped into a single catchment. This makes sense from our previous lineage/variant analysis: all our samples were classfied as Omicron VOC. We can see that our samples seem to be more diverged from the Australian BA.1 sample present in the background data, with new SNPs in our samples creating a longer branch in the tree.\n\n\n\nResults from civet report for catchment 1, showing the phylogeny in the context of a BA.1 Australian sample from the background data.\n\n\nCivet also outputs a CSV file (results/civet/master_metadata.csv), which includes the catchment that each sample was assigned to. We will use this CSV file later to integrate this information with other parts of our analysis, in R, detailed in the “Integration & Visualisation” section."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#integration-visualisation",
+ "href": "materials/03-case_studies/02-southafrica.html#integration-visualisation",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.7 Integration & Visualisation",
+ "text": "9.7 Integration & Visualisation\nAt this point in our analysis, we have several tables with different pieces of information:\n\nsample_info.csv → the original table with metadata for our samples.\nresults/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.\nresults/nextclade/nextclade.tsv → the results from Nextclade.\nresults/pangolin/pango_report.csv → the results from Pangolin.\nresults/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.\n\nTo consolidate our analysis, we tidied and integrated the information from across these different files, into a single table using the software R. The script used to do this is in scripts/07-data_integration.R. Because this is an R script, we opened it in RStudio to execute the code.\nThe output of our script is a new tab-delimited table, which we saved in report/consensus_metrics.tsv, and contains the following columns:\n\nsample → sample ID.\ncollection_date → date of collection day.\ncollection_week → date of collection week (useful for summarising/visualising counts per-week).\ncountry → country of origin.\nlatitude/longitude → latitude and longitude of collection.\nn_mapped_reads → number of mapped reads.\nmedian_depth → median depth.\npct_missing → percentage of missing data.\npct_coverage → percentage of coverage.\nn_variants → number of SNP + indel variants detected.\nnextclade → nextclade clade.\nqc_status → QC status as determined by Nextclade (“bad”, “mediocre”, “good”).\nlineage → Pangolin lineage.\nwho_variant → variant of concern designation.\ncatchment → catchment group from Civet.\n\nThis table, which aggregates information from many of the tools we used, was then used to produce different visualisations of our analysis. These visualisations were also done using the R software (scripts/08-visualisation.R), and integrated into a report, shown below."
+ },
+ {
+ "objectID": "materials/03-case_studies/02-southafrica.html#bonus-full-workflow",
+ "href": "materials/03-case_studies/02-southafrica.html#bonus-full-workflow",
+ "title": "9 South Africa (Illumina)",
+ "section": "9.8 Bonus: Full Workflow",
+ "text": "9.8 Bonus: Full Workflow\nAlthough we have ran each of the steps of our analysis individually (each in their own script), now that we have everything working, we could integrate all these steps into a single “master” script:\n#!/bin/bash\n\n# make mamba activate command available\neval \"$(conda shell.bash hook)\"\nsource $(mamba info --base)/etc/profile.d/mamba.sh\n\n# make report directory\nmkdir -p report\n\nmamba activate nextflow\n# run viralrecon\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '15.GB' --max_cpus 8 \\\n --platform illumina \\\n --input samplesheet.csv \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version 3 \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\n\n# combine and clean FASTA files\ncat results/viralrecon/variants/ivar/consensus/bcftools/*.consensus.fa | sed 's| MN908947.3||' > report/consensus.fa\n\nmamba activate seqkit\n# create missing bases TSV file\nseqkit locate -i -P -G -M -r -p \"N+\" report/consensus.fa > results/missing_intervals.tsv\n\nmamba activate nextclade\n# get nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade report/consensus.fa\n\nmamba activate pangolin\n# update pangolin data\npangolin --update-data\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv report/consensus.fa\n\nmamba activate phylo\n# alignment\nmkdir -p results/mafft\nmafft --6merpair --maxambiguous 0.2 --addfragments report/consensus.fa resources/reference/sarscov2.fa > results/mafft/alignment.fa\n\n# tree inference\nmkdir -p results/iqtree\niqtree2 -s results/mafft/alignment.fa --prefix results/iqtree/consensus\n\n# data integration and cleaning\nRscript scripts/07-data_integration.R\nNotice that we included the R script that does the data cleaning here, using the Rscript program that allows to execute an R script from the command-line.\nHaving this “master” script, we could run all these steps from start-to-finish with a single command, which can be very useful if you want to fully automate your analysis across multiple runs."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#pipeline-overview",
+ "href": "materials/03-case_studies/03-eqa.html#pipeline-overview",
+ "title": "10 EQA (Exercise)",
+ "section": "10.1 Pipeline Overview",
+ "text": "10.1 Pipeline Overview\nWe will start our analysis with FASTQ files produced by our sequencing platforms (Illumina and Nanopore are considered here). These FASTQ files will be used with the nf-core/viralrecon Nextflow pipeline, allowing us to automate the generation of consensus sequences and produce several quality control metrics essential for our downstream analysis and reporting.\nCritical files output by the pipeline will need to be further processed, including combining and cleaning our consensus FASTA files. Using these clean files, we can then proceed to downstream analysis, which includes assigning each sample to the most up-to-date Pango lineage, Nextclade clade and WHO designation. Finally, we can do more advanced analysis, including the idenfication of sample clusters based on phylogenetic analysis, or produce timeseries visualisations of variants of concern. With all this information together, we will have the necessary pieces to submit our results to public repositories and write reports to inform public health decisions."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#preparing-files",
+ "href": "materials/03-case_studies/03-eqa.html#preparing-files",
+ "title": "10 EQA (Exercise)",
+ "section": "10.2 Preparing Files",
+ "text": "10.2 Preparing Files\nBefore we start our work, it’s always a good idea to setup our directory structure, so we keep our files organised as the analysis progresses. There is no standard way to do this, but here are some suggested directories:\n\ndata → contains the sequencing data for the particular run or project you are working on. Data may sometimes be stored in a separate server, in which case you may not need to create this directory. Generally, you should leave the original data unmodified, in case something goes wrong during your analysis, you can always re-run it from the start.\nresults → to save results of the analysis.\nscripts → to save scripts used to run the analysis. You should always try to save all the commands you run in a script, this way you will have a record of what was done to produce the results, and it makes your life easier if you want to run the analysis again on a new set of data.\nreport → to save the final files and documents that we report to our colleagues or upload to public repositories.\nresources → files that you download from the internet could be saved here. For example, the reference genome sequence, background data used for phylogenetics, etc. Sometimes you may share these resources across multiple projects, in which case you could have this folder somewhere else that you can access across multiple projects.\n\nOn our computers, we have a directory in ~/Documents/eqa_workshop, where we will do all our analysis. We already include the following:\n\ndata → with the results of the EQA sample sequencing.\nresources → where we include the SARS-CoV-2 reference genome, and some background datasets that will be used with some of the tools we will cover.\nscripts → where we include some scripts that we will use towards the end of the workshop. You should also create several scripts during the workshop, which you will save here.\nsample_info.csv → a table with some metadata for our samples.\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nYour first task is to create two new directories in the project folder called report and results. You can do this either using the file explorer or from the command line (using the mkdir command).\n\n\n\n\n\n\n10.2.1 Data\nRegardless of which platform you used to sequence your samples, the analysis starts with FASTQ files (if you need a reminder of what a FASTQ file is, look at the Introduction to NGS section). However, the organisation of these files is slightly different depending on the platform, and is detailed below.\n\nNanoporeIllumina\n\n\nTypically, Nanopore data is converted to FASTQ format using the program Guppy. This software outputs the files to a directory called fastq_pass. Within this directory, it creates further sub-directories for each sample barcode, which are named barcodeXX (XX is the barcode number). Finally, within each barcode directory there is one (or sometimes more) FASTQ files corresponding to that sample.\nYou can look at the files you have available from the command line using:\nls data/fastq_pass\n\n\nThe Illumina files come as compressed FASTQ files (.fq.gz format) and there’s two files per sample, corresponding to read 1 and read 2. This is indicated by the file name suffix:\n\n*_1.fq.gz for read 1\n*_2.fq.gz for read 2\n\nYou can look at the files you have available from the command line using:\nls data/\n\n\n\n\n\n10.2.2 Metadata\nA critical step in any analysis is to make sure that our samples have all the relevant metadata associated with them. This is important to make sense of our results and produce informative reports at the end. There are many types of information that can be collected from each sample (revise the Genomic Surveillance > Metadata section of the materials to learn more about this). For effective genomic surveillance, we need at the very minimum three pieces of information:\n\nWhen: date when the sample was collected (not when it was sequenced!).\nWhere: the location where the sample was collected (not where it was sequenced!).\nHow: how the sample was sequenced (sequencing platform and protocol used).\n\nOf course, this is the minimum metadata we need for a useful analysis. However, the more information you collect about each sample, the more questions you can ask from your data – so always remember to record as much information as possible for each sample.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nNote: If you are using our pre-sequenced data, you can skip this exercise.\nWe already provide some basic metadata for these samples in the file sample_info.csv:\n\nsample → the sample ID.\ncollection_date → the date of collection for the sample in the format YYYY-MM-DD.\ncountry → the country of origin for this sample.\nexpected_lineage and expected_voc → because we are using the EQA panel, we know what lineage and variant-of-concern (VOC) each sample belongs to. We will use this later to assess the quality of our analysis.\n\nThere is however some information missing from this table, which you may have available at this point. Open this file in Excel and create the following columns:\n\nct → Ct value from qPCR viral load quantification.\nsequencing_instrument → the model for the sequencing instrument used (e.g. NovaSeq 6000, MinION, etc.).\nsequencing_protocol_name → the type of protocol used to prepare the samples (e.g. ARTIC).\namplicon_primer_scheme → for amplicon protocols, what version of the primers was used (e.g. V3, V4.1)\nSpecific columns for Oxford Nanopore data, which are essential for the bioinformatic analysis:\n\nont_nanopore → the version of the pores used (e.g. 9.4.1 or 10.4.1).\nont_guppy_version → the version of the Guppy software used for basecalling.\nont_guppy_mode → the basecalling mode used with Guppy (usually “fast”, “high”, “sup” or “hac”).\n\n\nThis will ensure that in the future people have sufficient information to re-run the analysis on your data.\n\n\n\n\n\n\n\n\n\n\n\nDates in Spreadsheet Programs\n\n\n\nNote that programs such as Excel often convert date columns to their own format, and this can cause problems when analysing data later on. For example, GISAID wants dates in the format YYYY-MM-DD, but by default Excel displays dates as DD/MM/YYYY.\nYou can change how Excel displays dates by highlighting the date column, right-clicking and selecting Format cells, then select “Date” and pick the format that matches YYYY-MM-DD. However, every time you open the CSV file, Excel annoyingly converts it back to its default format!\nTo make sure no date information is lost due to Excel’s behaviour, it’s a good idea to store information about year, month and day in separate columns (stored just as regular numbers)."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#consensus-assembly",
+ "href": "materials/03-case_studies/03-eqa.html#consensus-assembly",
+ "title": "10 EQA (Exercise)",
+ "section": "10.3 Consensus Assembly",
+ "text": "10.3 Consensus Assembly\nAt this point we are ready to start our analysis with the first step: generating a consensus genome for our samples. We will use a standardised pipeline called viralrecon, which automates most of this process for us, helping us be more efficient and reproducible in our analysis.\n\n\n\n\n\n\nNote\n\n\n\nSee Section 4.1, if you need to revise how the nf-core/viralrecon pipeline works.\n\n\n\n10.3.1 Samplesheet\nThe first step in this process is to prepare a CSV file with information about our sequencing files, which will be used as the input to the viralrecon pipeline.\nThe pipeline’s documentation gives details about the format of this samplesheet, depending on whether you are working with Illumina or Nanopore data.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nNote: If you are using our pre-sequenced data, you can skip this exercise.\nUsing Excel, produce the input samplesheet for nf-core/viralrecon, making sure that you save it as a CSV file (File → Save As… and choose “CSV” as the file format).\nNote: make sure that the sample names you use in this samplesheet match those in the metadata sample_info.csv file (pay attention to things like spaces, uppercase/lowercase, etc. – make sure the names used match exactly).\nIf you are working with Illumina data, you should check the tip below.\n\n\nIllumina samplesheet: saving time with the command line!\n\nYou can save some time (and a lot of typing!) in making the Illumina samplesheet using the command line to get a list of file paths. For example, if your files are saved in a folder called data, you could do:\n# list read 1 files and save output in a temporary file\nls data/*_1.fq.gz > read1_filenames.txt\n\n# list read 2 files and save output in a temporary file\nls data/*_2.fq.gz > read2_filenames.txt\n\n# initiate a file with column names\necho \"fastq_1,fastq_2\" > samplesheet.csv\n\n# paste the two temporary files together, using comma as a delimiter\npaste -d \",\" read1_filenames.txt read2_filenames.txt >> samplesheet.csv\n\n# remove the two temporary files\nrm read1_filenames.txt read2_filenames.txt\nNow, you can open this file in Excel and continue editing it to add a new column of sample names.\n\n\n\n\n\n\n\n\n10.3.2 Running Viralrecon\nThe next step in our analysis is to run the nf-core/viralrecon pipeline. The way the command is structured depends on which kind of data we are working with. There are many options that can be used to customise the pipeline, but typical commands are shown below for each platform.\n\nNanoporeIllumina\n\n\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '15.GB' --max_cpus 4 \\\n --platform nanopore \\\n --input SAMPLESHEET_CSV \\\n --fastq_dir data/fastq_pass/ \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version PRIMER_VERSION \\\n --artic_minion_caller medaka \\\n --artic_minion_medaka_model MEDAKA_MODEL \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\nYou need to check which model the medaka software should use to process the data. You need three pieces of information to determine this:\n\nThe version of the nanopores used (usually 9.4.1 or 10.4.1).\nThe sequencing device model (MinION, GridION or PromethION).\nThe mode used in the Guppy software for basecalling (“fast”, “high”, “sup” or “hac”).\nThe version of the Guppy software.\n\nOnce you have these pieces of information, you can see how the model is specified based on the model files available on the medaka GitHub repository. For example, if you used a flowcell with chemistry 9.4.1, sequenced on a MinION using the “fast” algorithm on Guppy version 5.0.7, the model used should be r941_min_fast_g507.\nNote that in some cases there is no model for recent versions of Guppy, in which case you use the version for the latest version available. In our example, if our version of Guppy was 6.1.5 we would use the same model above, since that’s the most recent one available.\n\n\nnextflow run nf-core/viralrecon \\\n -r 2.6.0 -profile singularity \\\n --max_memory '15.GB' --max_cpus 4 \\\n --platform illumina \\\n --input SAMPLESHEET_CSV \\\n --outdir results/viralrecon \\\n --protocol amplicon \\\n --genome 'MN908947.3' \\\n --primer_set artic \\\n --primer_set_version PRIMER_VERSION \\\n --skip_assembly --skip_asciigenome \\\n --skip_pangolin --skip_nextclade\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nYour next task is to run the pipeline on your data. However, rather than run the command directly from the command line, let’s save it in a shell script – for reproducibility and as a form of documenting our analysis.\n\nFirst, open the README.txt file found in your folder, which has some details about your samples, including the amplicon primer scheme used.\nUsing a text editor, create a new shell script and save it in scripts/01-run_viralrecon.sh. You can either use the command-line text editor nano or use Gedit, which comes installed with Ubuntu.\nCopy the command shown above (either Illumina or Nanopore, depending on your data) to your new script.\nAdjust the code, to use the correct input samplesheet file, primer scheme and, in the case of ONT, the medaka model. For information about primer scheme options see the “Amplicon Primer Schemes” appendix.\nActivate the software environment to use Nextflow: mamba activate nextflow.\nSave the script and run it from the command line using bash scripts/01-run_viralrecon.sh.\n\nIf you need a reminder of how to work with shell scripts, revise the Shell Scripts section of the accompanying Unix materials.\n\n\n\n\n\n\n\n\n\n\n\nMaximum Memory and CPUs\n\n\n\nIn our Nextflow command above we have set --max_memory '15.GB' --max_cpus 8 to limit the resources used in the analysis. This is suitable for the computers we are using in this workshop. However, make sure to set these options to the maximum resources available on the computer where you process your data."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#consensus-quality",
+ "href": "materials/03-case_studies/03-eqa.html#consensus-quality",
+ "title": "10 EQA (Exercise)",
+ "section": "10.4 Consensus Quality",
+ "text": "10.4 Consensus Quality\nOnce your workflow is complete, it’s time to assess the quality of the assembly.\n\n\n\n\n\n\nNote\n\n\n\nSee Section 5.2, if you need to revise how to assess the quality of consensus sequences.\n\n\n\n10.4.1 Coverage\nAt this stage we want to identify issues such as:\n\nAny samples which have critically low coverage. There is no defined threshold, but samples with less than 85% coverage should be considered carefully.\nAny problematic regions that systematically did not get amplified (amplicon dropout).\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nTo assess the quality of our assemblies, we can use the MultiQC report generated by the pipeline, which compiles several pieces of information about our samples. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.\nOpen the quality report and try to answer the following questions:\n\nWere there any samples with more than 15% missing bases (’N’s)?\nWere there any samples with a median depth of coverage <20x?\nWere there any problematic amplicons with low depth of coverage across multiple samples?\n\nMake a note of any samples that you think are problematic. You can discuss with your colleagues and compare your results/conclusions to see if you reach similar conclusions.\n\n\n\n\n\n\n\n10.4.2 Variants\nThe viralrecon pipeline outputs a table with information about SNP/Indel variants as a CSV file named variants_long_table.csv. It is important to inspect the results of this file, to identify any mutations with severe effects on annotated proteins, or identify samples with an abnormal high number of “mixed” bases.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nOpen the variants_long_table.csv file and answer the following questions. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.\n\nHow many variants have allele frequency < 75%?\nDoes any of the samples have a particularly high number of these low-frequency variants, compared to other samples? (This could indicate cross-contamination of samples)\nInvestigate if there are any samples with frameshift mutations. These mutations should be rare because they are highly disruptive to the functioning of the virus. So, their occurrence may be due to errors rather than a true mutation and it’s good to make a note of this.\n\nIf you need a reminder of the meaning of the columns in this file, consult the Consensus > Mutation/Variant Analysis section of the materials.\nMake a note of any samples that you think may be problematic, either because they have a high number of low-frequency variants or because they have frameshift mutations.\n(Optional)\nIf you identify samples with frameshift mutations, open the BAM file of the sample using the software IGV. Go to the position where the mutation is located, and try to see if there is evidence that this mutation is an error (for example, if it’s near an homopolymer).\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nThe samples we are using in this analysis come from a standard EQA panel. As such, we already know what lineages we expect to find in these samples.\n\nOpen IGV and load one (or more) of the alignment BAM files into it. If you need a reminder of where to find this file, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.\nOpen your metadata table (sample_info.csv) and check which lineages your loaded samples belong to.\nDo a web search to find what mutations characterise those lineages.\nOn IGV, go to the location of some of the expected mutations, and see if you can see it in the respective samples.\n\n\n\n\n\n\n\n\n10.4.3 Clean FASTA\nThe viralrecon pipeline outputs each of our consensus sequences as individual FASTA files for each sample (look at the FASTA Files section if you need a reminder of what these files are). However, by default, the sample names in this file have extra information added to them, which makes some downstream analysis less friendly (because the names will be too long and complicated).\nTo clean up these files, we will do two things:\n\nCombine all the individual FASTA files together.\nRemove the extra text from the sequence names.\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nWe will use the programs cat (concatenate) and sed (text replacement) to clean our FASTA files. Consider the commands given below:\n\nNanoporeIllumina\n\n\ncat <INPUT> | sed 's|/ARTIC/medaka MN908947.3||' > report/consensus.fa\n\n\ncat <INPUT> | sed 's| MN908947.3||' > report/consensus.fa\n\n\n\nThe sed command shown is used to remove the extra pieces of text added by the pipeline to each sample name. This is a slightly more advanced program to use, so we already give you the code. If you want to learn more about it, check the “Text Replacement” section of the accompanying Unix materials.\nCopy the command above to a new shell script called scripts/02-clean_fasta.sh. Fix the code by replacing <INPUT> with the path to all the FASTA files generated by the pipeline (remember you can use the * wildcard).\nIf you need a reminder of where to find the FASTA consensus files from viralrecon, consult the “Consensus assembly” section of the materials, or the Viralrecon output documentation.\nOnce you fixed the script, run it with bash and check that the output file is generated.\nBonus:\nCheck that you have all expected sequences in your FASTA file using grep to find the lines of the file with > (which is used to indicate sample names).\n\n\n\n\n\n\n\n10.4.4 Missing Intervals\nWhen we generate our consensus assembly, there are regions for which we did not have enough information (e.g. due to amplicon dropout) or where there were mixed bases. These regions are missing data, and are denoted as ‘N’ in our sequences. Although we already determined the percentage of each sequence that is missing from the MultiQC report, we don’t have the intervals of missing data, which can be a useful quality metric to have. In particular, we may want to know what is the largest continuous stretch of missing data in each sample.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nWe will use the software tool seqkit to find intervals where the ‘N’ character occurs. This tool has several functions available, one of which is called locate.\nHere is the command that would be used to locate intervals containing one or more ‘N’ characters:\nseqkit locate -i -P -G -M -r -p \"N+\" report/consensus.fa\nThe meaning of the options is detailed in seqkit’s documentation.\n\nCreate a new shell script called scripts/03-missing_intervals.sh.\nCopy the command shown above to the script and modify it to redirect the output (using >) to a file called results/missing_intervals.tsv.\nActivate the software environment: mamba activate seqkit.\nRun the script you created using bash.\n\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nOpen the file you created in the previous step (results/missing_intervals.tsv) in a spreadsheet program. Create a new column with the length of each interval (end - start + 1).\nNote if any missing intervals are larger than 1Kb, and whether they overlap with the Spike gene. (Note: you can use the Sars-CoV-2 genome browser to help you see the location of the annotated genes.)"
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#downstream-analyses",
+ "href": "materials/03-case_studies/03-eqa.html#downstream-analyses",
+ "title": "10 EQA (Exercise)",
+ "section": "10.5 Downstream Analyses",
+ "text": "10.5 Downstream Analyses\nNow that we have cleaned our FASTA file, we can use it for several downstream analysis. We will focus on these:\n\nLineage assignment: identify if our samples come from known lineages from the Pangolin consortium.\nPhylogeny: produce an annotated phylogenetic tree of our samples.\nClustering: assess how many clusters of sequences we have, based on a phylogenetic analysis.\nIntegration & Visualisation: cross-reference different results tables and produce visualisations of how variants changed over time.\n\n\n10.5.1 Lineage Assignment\n\n\n\n\n\n\nNote\n\n\n\nSee Section 6.1, if you need to revise how lineage assignment works.\n\n\nAlthough the Viralrecon pipeline can run Pangolin and Nextclade, it does not use the latest version of these programs (because lineages evolve so fast, the nomenclature constantly changes). Although it is possible to configure viralrecon to use more recent versions of these tools, it requires more advanced use of configuration files with the pipeline.\nAlternatively, we can run our consensus sequences through the latest versions of Nextclade and Pangolin. There are two ways to do this: using their respective web applications, or their command-line versions. For this task, we recommend that you use the command line tools (this will ensure our downstream analysis works well), but we also provide the option to use the web apps for your reference.\n\nCommand lineWeb Apps\n\n\nTo run the command-line version of these tools, there are two steps:\n\nUpdate the datasets of each software (to ensure they are using the latest lineage/clade nomenclature available).\nRun the actual analysis on your samples.\n\nWe will use the exercises below to see what the commands to achieve this are.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nWe will start doing the Nextclade analysis.\n\nCreate a new script file called scripts/04-nextclade.sh and copy this code into it:\n# update nextclade data\nnextclade dataset get --name sars-cov-2 --output-dir resources/nextclade_background_data\n\n# run nextclade\nnextclade run --input-dataset resources/nextclade_background_data/ --output-all results/nextclade/ <INPUT>\nFix the code, replacing <INPUT> with the path to your consensus sequence file. Save the file.\nActivate the software environment: mamba activate nextclade.\nRun your script using bash.\nOnce the analysis completes, open the file results/nextclade/nextclade.tsv in Excel and see what problems your samples may have (in particular those classified as “bad” quality).\n\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nFor the Pangolin analysis:\n\nCreate a new script file called scripts/04-pangolin.sh and copy this code into it:\n# update pangolin data\n<DATA_UPDATE_COMMAND>\n\n# run pangolin\npangolin --outdir results/pangolin/ --outfile pango_report.csv <INPUT>\nFix the code:\n\nReplace <INPUT> with the path to your consensus sequence file.\nReplace <DATA_UPDATE_COMMAND> with the pangolin command used to update its data. Check the documentation of the tool with pangolin --help to see if you can find what this option is called. Alternatively, look at the documentation online.\n\nSave the file.\nActivate the software environment: mamba activate pangolin.\nRun your script using bash.\nOnce the analysis completes, open the file results/pangolin/pango_report.csv in Excel and see if there were any samples for which the analysis failed. If there were any failed samples, check if they match the report from Nextclade.\n\n\n\n\n\n\n\n\nThese exercises are optional - during the workshop please use the command line version of the tools.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nRunning Nextclade\nGo to clades.nextstrain.org and run Nextclade on the clean FASTA file you created earlier (report/consensus.fa).\nIf you need a reminder about this tool, see the “Lineages and variants” section of the materials.\nOnce the analysis completes, pay particular attention to the quality control column, to see what problems your samples may have (in particular those classified as “bad” quality).\nThen:\n\nCreate a new folder in your project directory: results/nextclade.\nUse the “download” button (top-right) and download the file nextclade.tsv (tab-delimited file), which contains the results of the analysis. Important: please download the TSV file (not the CSV file, as it uses ; to separate columns, and will not work later with the “Data Integration” section).\nSave this file in the results/nextclade folder you created.\n\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nRunning Pangolin\nGo to pangolin.cog-uk.io and run Pangolin on the clean FASTA file you created earlier (report/consensus.fa).\nIf you need a reminder about this tool, see the “Lineages and variants” section of the materials.\nOnce the analysis completes, pay particular attention to any samples that failed. If there were any failed samples, check if they match the report from Nextclade.\nThen:\n\nCreate a new folder in your project directory: results/pangolin.\nUse the “download” button (the “arrow down” symbol on the results table) and download the file results.csv. Rename this file to report.csv (important: make sure to rename the file like this, otherwise it will not work later with the “Data Integration” section).\nSave this file in the results/pangolin folder you created.\n\n\n\n\n\n\n\n\n\n\n\n10.5.2 Phylogeny\n\n\n\n\n\n\nNote\n\n\n\nSee Section 7.1, if you need to revise how to build phylogenetic trees.\n\n\nAlthough tools such as Nextclade and civet can place our samples in a phylogeny, sometimes it may be convenient to build our own phylogenies. This requires three steps:\n\nProducing a multiple sequence alignment from all consensus sequences.\nTree inference.\nTree visualisation.\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\n\nStart by creating two directories to store the output of our analysis: results/mafft and results/iqtree.\nActivate the software environment: mamba activate phylo (this environment includes both mafft and iqtree).\nTo make our analysis more interesting, we will combine our sequences with sequences from previous workshops. Use the cat program to concatenate your sequences (report/consensus.fa) with the sequences from our collaborators (resources/eqa_collaborators/eqa_consensus.fa). Using >, save the output in a new file results/mafft/unaligned_consensus.fa.\nPerform a multiple sequence alignment of the combined consensus sequences using the program mafft (see Section 7.2 for how to use this program). Save the output in a file called results/mafft/aligned_consensus.fa.\nInfer a phylogenetic tree using the iqtree2 program (see Section 7.3).\nOnce you have both of these commands working, make sure to save them in a new shell script (as a record of your analysis). Save the script as scripts/05-phylogeny.sh.\nVisualise the tree using FigTree (see Section 7.4).\n\nWhat substitution model was chosen as the best for your data by IQ-Tree?\nDo your samples group with samples from previous workshops in the way that is expected, or are there any outliers?\nAre there any obvious differences between sequencing technologies?\n\n\n\n\n\n\n\n10.5.3 Clustering (Optional Section)\n\nThe software civet (Cluster Investigation and Virus Epidemiology Tool) can be used to produce a report on the phylogenetic relationship between viral samples, using a background dataset to contextualise them (e.g. a global or regional sample of sequences) as well as metadata information to enable the identification of emerging clusters of samples.\nCivet works in several stages, in summary:\n\nEach input sequence is assigned to a “catchment” group. These are groups of samples that occur at a given genetic distance (number of SNP differences) from each other.\nWithin each catchment group, a phylogeny is built (using the iqtree2 software with the HKY substitution model).\nThe phylogeny is annotated with user-input metadata (e.g. location, collection date, etc.). The results are displayed in an interactive HTML report.\n\nOne of the critical pieces of information needed by civet is the background data, to which the input samples will be compared with. These background data could be anything the user wants, but typically it’s a good idea to use samples from the local geographic area from where the samples were collected. For example, a national centre may choose to have their background data composed of all the samples that have been sequenced in the country so far. Or perhaps all the samples in its country and other neighbouring countries.\nOne way to obtain background data is to download it from GISAID. An example of how to do this is detailed in the civet documentation. We have already downloaded the data from GISAID, so we can go straight to running our civet analysis.\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\n\nCreate a new script in scripts/06-civet.sh, with the following command, adjusted to fit your files:\ncivet -i <PATH_TO_YOUR_SAMPLE_METADATA> \\\n -f <PATH_TO_YOUR_CONSENSUS_FASTA> \\\n -icol <COLUMN_NAME_FOR_YOUR_SAMPLE_IDS> \\\n -idate <COLUMN_NAME_FOR_YOUR_COLLECTION_DATE> \\\n -d <PATH_TO_CIVET_BACKGROUND_DATA> \\\n -o results/civet\nActivate the software environment: mamba activate civet.\nOnce your script is ready, run it with bash.\nAfter the analysis completes, open the HTML output file in results/civet and examine into how many catchments your samples were grouped into."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#integration-visualisation",
+ "href": "materials/03-case_studies/03-eqa.html#integration-visualisation",
+ "title": "10 EQA (Exercise)",
+ "section": "10.6 Integration & Visualisation",
+ "text": "10.6 Integration & Visualisation\nAt this point in our analysis, we have several tables with different pieces of information:\n\nsample_info.csv → the original table with metadata for our samples.\nresults/viralrecon/multiqc/medaka/summary_variants_metrics_mqc.csv → quality metrics from the MultiQC report generated by the viralrecon pipeline.\nresults/nextclade/nextclade.tsv → the results from Nextclade.\nresults/pangolin/pango_report.csv → the results from Pangolin.\n(optional) results/civet/master_metadata.csv → the results from the civet analysis, namely the catchment (or cluster) that each of our samples was grouped into.\n\nEach of these tables stores different pieces of information, and it would be great if we could integrate them together, to facilitate their interpration and generate some visualisations.\nWe will demonstrate how this analysis can be done using the R software, which is a popular programming language used for data analysis and visualisation. Check the Quick R Intro appendix for the basics of how to work with R and RStudio.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nData Integration\nFrom RStudio, open the script in scripts/07-data_integration.R. Run the code in the script, going line by line (remember in RStudio you can run code from the script panel using Ctrl + Enter). As you run the code check the tables that are created (in your “Environment” panel on the top-right) and see if they were correctly imported.\nOnce you reach the end of the script, you should have two tab-delimited files named report/consensus_metrics.tsv and report/variants.tsv. Open both files in Excel to check their content and confirm they contain information from across the multiple tables.\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nData Visualisation\nAfter integrating the data, it’s time to produce some visualisations. From RStudio, open the script in scripts/08-visualisation.R. Run the code in the script, going line by line.\nAs you run the code, several plots will be created. You can export these plots from within RStudio using the “Export” button on the plotting panel – these will be useful when you write your report later.\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nAnnotating Phylogenetic Tree\nUsing FigTree, import two annotation files (File > Import Annotations…):\n\nreport/consensus_metrics.tsv, which was created in the Data Integration exercise.\nresources/eqa_collaborators/metadata.tsv, which has lineage assignment for EQA samples sequenced by other labs.\n\nAfter importing both files, annotate your phylogenetic tree to display the lineages assigned to each sample as the tip labels. See Section 7.4, if you need a reminder of how to annotate trees using FigTree."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#eqa-panels",
+ "href": "materials/03-case_studies/03-eqa.html#eqa-panels",
+ "title": "10 EQA (Exercise)",
+ "section": "10.7 EQA Panels",
+ "text": "10.7 EQA Panels\nThe analysis we have done so far is applicable to any new sequences that you want to process. However, we have also been using a pre-defined panel of samples to be used as part of an External Quality Assessment (EQA) process. This allows us to assess whether our bioinformatic analysis identified all the expected mutations in these samples, as well as assigned them to the correct lineages.\nTo do this, requires to compare the mutations we detected in our EQA samples to the expected mutations provided to us. From this, we will be able to calculate True Positives (TP), False Positives (FP) and False Negatives (FN), and calculate two performance metrics:\n\nPrecision = TP / (TP + FP) → what fraction of the mutations we detected are true?\nSensitivity = TP / (TP + FN) → what fraction of all true mutations did we detect?\n\nThere can be cases where one of these metrics is high and the other one lower. For example, if you have a high-coverage genome (say >95%) but lots of sequencing errors, you may have a high sensitivity (you manage to detect all true mutations), but low precision (you also detect lots of false positives, due to sequencing errors). Conversely, if you have a low-coverage genome (say <50%) but very high-quality sequencing, you may have low sensitivity (you missed lots of true mutations, because of missing data), but high precision (those mutations that you did manage to identify were all true, you didn’t have many false positives).\n\nIf you are submiting your samples to GenQA’s platform, they will also provide with a general accuracy score called F-score. This is calculated as the harmonic mean of precision and sensitivity:\n\\(F_{score} = \\frac{2 \\times Precision \\times Sensitivity}{Precision + Sensitivity}\\)\nA high F-score is indicative of both high precision and sensitivity, whereas a lower score indicates that at least one of those metrics are low.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nFrom RStudio, open the script in scripts/09-eqa_validation.R. Run the code in the script, going line by line.\nThis script will save a new tab-delimited file in report/eqa_performance_metrics.tsv. Open this file in Excel and check what the precision and sensitivity of your analysis was. Are you happy with the results?\n\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\n(Optional)\nWhile you run the R script, you will have an object called all_mutations in your environment (check the top-right panel). From RStudio, click on the name of this object to open it in the table viewer.\nLook at mutations that were not detected in your samples, but were present in the expected mutations (i.e. false negatives). Open the BAM file for one of those samples in IGV and navigate to the position where that mutation should occur. Investigate what the reason may have been for missing that mutation.\nIf you need a reminder of where to find the BAM file, consult the Consensus > Output Files section of the materials, or the Viralrecon output documentation."
+ },
+ {
+ "objectID": "materials/03-case_studies/03-eqa.html#reporting",
+ "href": "materials/03-case_studies/03-eqa.html#reporting",
+ "title": "10 EQA (Exercise)",
+ "section": "10.8 Reporting",
+ "text": "10.8 Reporting\nFinally, we have come to the end of our analysis. So, all that is left is to report our results in the most informative way to our colleagues and decision-makers. You may already have established reporting templates in your institution, and if that’s the case, you should use those. Alternatively, we will look at a suggested template, based on the reports done by the UK Health Security Agency (UKHSA).\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nOpen our shared report template document and download it as a Word document (File → Download → Microsoft Word (.docx)). Save the file in report/YYYY-MM-DD_analysis_report.docx (replace YYYY-MM-DD by today’s date).\nOpen the file and complete it with the information you collected throughout your analysis. You should be able to get all this information from the files in your report/ directory."
+ },
+ {
+ "objectID": "materials/04-wastewater/00-ww_temp.html",
+ "href": "materials/04-wastewater/00-ww_temp.html",
+ "title": "11 Wastewater surveillance",
+ "section": "",
+ "text": "Caution\n\n\n\nUnder development"
+ },
+ {
+ "objectID": "materials/05-software/01-managing_software.html#why-linux",
+ "href": "materials/05-software/01-managing_software.html#why-linux",
+ "title": "12 Managing Bioinformatics Software",
+ "section": "12.1 Why Linux?",
+ "text": "12.1 Why Linux?\nMost computer users either use Windows or macOS as their operating system. However, in bioinformatics the Linux operating system is the most popular.\nThere are different reasons to prefer Linux, for example:\n\nMost bioinformatics software is only available for this operating system.\nGenerally more secure.\nFree and open source.\nThe operating system of choice on high performance compute (HPC) cluster environments.\nIt has flexible built-in command line tools to manipulate large data (cat, grep, sed, and the | pipe for chaining commands).\n\nBecause Linux is an open source project, there are many different types of Linux operating systems available. Generally, we recommend using Ubuntu, as it’s a well-supported and widely used distribution.\nThe first step for running bioinformatics software therefore is to ensure that you have access to Ubuntu Linux on your machine. We give instructions for this in Section 13.1."
+ },
+ {
+ "objectID": "materials/05-software/01-managing_software.html#package-managers",
+ "href": "materials/05-software/01-managing_software.html#package-managers",
+ "title": "12 Managing Bioinformatics Software",
+ "section": "12.2 Package Managers",
+ "text": "12.2 Package Managers\nOnce you have Linux available, there is the question of how to install software, especially when using the command line. Most modern Linux operating systems have a default package manager available. A package manager allows the user to install (or remove, or upgrade) software with a single command. The package manager takes care of automatically downloading and installing the software we want, as well as any dependencies it requires.\n\n\n\nDiagram illustrating how package managers work. Image source.\n\n\n\n12.2.1 Ubuntu apt\nOn Ubuntu, the default package manager is called apt. This software can be used to do system-wide updates, upgrades and installation of software packages. There are several commands available:\n\napt update updates the database of available packages, to check the latest versions available. This command doesn’t actually install anything.\napt upgrade upgrades all the packages to their latest version. This command upgrades existing packages, and installs new ones if required as dependencies.\napt install is used to install a single software of your choice.\n\nFor example, let’s say we wanted to install the latest version of java (Java is used for many applications, including IGV, which we introduced earlier). We could run the following commands:\nsudo apt update # make sure the package database is up-to-date\nsudo apt upgrade # upgrade existing software to their latest version\nsudo apt install default-jre # install java\nThe sudo command at the beginning indicates that we want to run the apt command with administrator privilege, which will require you to input your password. This is necessary, because the apt command does a system installation of the software, so it can only be done if the user trying to do the installation has those permissions. With the install command, we give the name of the software we want to install. In this case, default-jre will install the default Java runtime environment.\nThe apt package manager is extremely useful to install core system software, however most bioinformatics software are not available from apt. Also, as it requires administrator (sudo) permissions, it is not always possible to install software with apt (for example, if you are using a HPC cluster). This is why we turn to alternative package managers such as Conda.\n\n\n12.2.2 Debian Packages\nSometimes, software is not available through the apt repositories, but instead distributed as a file. Ubuntu uses the Debian package format. To install Debian packages, you can use the dpkg command.\nFor example, the RStudio application for Linux is distributed as a .deb file. After you download it, you can install it as follows:\nsudo dpkg -i rstudio-2023.12.0-369-amd64.deb\nAs with apt this is a system installation and so requires admin privileges (sudo).\n\n\n12.2.3 Conda/Mamba Package Manager\nOften you may want to use software packages that are not available on the apt repositories. A popular alternative in bioinformatics is to use the package manager Mamba, which is a successor to another package manager called Conda.\nConda and Mamba are package managers commonly used in data science, scientific computing, and bioinformatics. Conda, originally developed by Anaconda, is a package manager and environment manager that simplifies the creation, distribution, and management of software environments containing different packages and dependencies. It is known for its cross-platform compatibility and ease of use. Mamba is a more recent and high-performance alternative to Conda. While it maintains compatibility with Conda’s package and environment management capabilities, Mamba is designed for faster dependency resolution and installation, making it a better choice nowadays.\nOne of the strengths of using Mamba to manage your software is that you can have different versions of your software installed alongside each other, organised in environments. Organising software packages into environments is extremely useful, as it allows to have a reproducible set of software versions that you can use and reuse in your projects.\nFor example, imagine you are working on two projects with different software requirements:\n\nProject A: requires Python 3.7, NumPy 1.15, and scikit-learn 0.20.\nProject B: requires Python 3.9, the latest version of NumPy, and TensorFlow 2.0.\n\nIf you don’t use environments, you would need to install and maintain these packages globally on your system. This can lead to several issues:\n\nVersion conflicts: different projects may require different versions of the same library. For example, Project A might not be compatible with the latest NumPy, while Project B needs it.\nDependency chaos: as your projects grow, you might install numerous packages, and they could interfere with each other, causing unexpected errors or instability.\nDifficulty collaborating: sharing your code with colleagues or collaborators becomes complex because they may have different versions of packages installed, leading to compatibility issues.\n\n\n\n\nIllustration of Conda/Mamba environments. Each environment is isolated from the others (effectively in its own folder), so different versions of the packages can be installed for distinct projects or parts of a long analysis pipeline.\n\n\nEnvironments allow you to create isolated, self-contained environments for each project, addressing these issues:\n\nIsolation: you can create a separate environment for each project using tools like Conda/Mamba. This ensures that the dependencies for one project don’t affect another.\nVersion control: you can specify the exact versions of libraries and packages required for each project within its environment. This eliminates version conflicts and ensures reproducibility.\nEase of collaboration: sharing your code and environment file makes it easy for collaborators to replicate your environment and run your project without worrying about conflicts.\nSimplified maintenance: if you need to update a library for one project, it won’t impact others. You can manage environments separately, making maintenance more straightforward.\n\nAnother advantage of using Mamba is that the software is installed locally (by default in your home directory), without the need for admin (sudo) permissions.\nYou can search for available packages from the anaconda.org website. Packages are organised into “channels”, which represent communities that develop and maintain the installation “recipes” for each software. The most popular channels for bioinformatics and data analysis are “bioconda” and “conda-forge”.\nThere are three main commands to use with Mamba:\n\nmamba create -n ENVIRONMENT-NAME: this command creates a new software environment, which can be named as you want. Usually people name their environments to either match the name of the main package they are installating there (e.g. an environment called pangolin if it’s to install the Pangolin software). Or, if you are installing several packages in the same environment, then you can name it as a topic (e.g. an environment called rnaseq if it contains several packages for RNA-seq data analysis).\nmamba install -n ENVIRONMENT-NAME NAME-OF-PACKAGE: this command installs the desired package in the specified environment.\nmamba activate ENVIRONMENT-NAME: this command “activates” the environment, which means the software installed there becomes available from the terminal.\n\nLet’s see a concrete example. If we wanted to install packages for phylogenetic analysis, we could do:\n# create an environment named \"phylo\"\nmamba create -n phylo\n \n# install some software in that environment\nmamba install -n phylo iqtree mafft\nIf we run the command:\nmamba env list\nWe will get a list of environments we created, and “phylo” should be listed there. If we want to use the software we installed in that environment, then we can activate it:\nmamba activate phylo\nAnd usually this changes your terminal to have the word (phylo) at the start of your prompt."
+ },
+ {
+ "objectID": "materials/05-software/01-managing_software.html#software-containers",
+ "href": "materials/05-software/01-managing_software.html#software-containers",
+ "title": "12 Managing Bioinformatics Software",
+ "section": "12.3 Software Containers",
+ "text": "12.3 Software Containers\nSoftware containerization is a way to package software and its dependencies in a single file. A software container can be thought of as a very small virtual machine, with everything needed to run that software stored inside that file. For this reason, software containerization solutions, such as Docker and Singularity, are widely used in bioinformatics.\nSoftware containers ensure reproducibility, allowing the same analysis to run on different systems. They can run on a local computer or on a high-performance computing cluster, producing the same result. The software within a container is isolated from other software, addressing the issue of incompatible dependencies between tools (similarly to Mamba environments).\nAs we already saw, analysis pipelines can be very complex, using many tools, each with their own dependencies. Therefore, worflow managers such as Nextflow use software containers to run their analysis (both Singularity and Docker are supported). This is what we’ve been doing throughout these materials, when running the nf-core/viralrecon pipeline, where we used the option -profile singularity. With this option, Nextflow will download the necessary software containers to run each step of the pipeline, which are available in public repositories online.\nTo use Singularity and Docker containers, the respective programs have to be installed, which we detail in our software setup page.\n\n\n\n\n\n\nPackage managers or containers?\n\n\n\nAs we’ve seen, the Mamba package manager and the containerisation solutions such as Docker and Singularity are trying to achieve similar things: enabling the reproducible installation of software and its dependencies in an isolated environment. So, why use one or the other?\nMamba is more user-friendly, allowing you to easily install packages of your choice. However, Mamba often works less well for more complex environments and can become extremely inneficient for environments with too many packages and conflicting versions. The recommendation is to keep your Mamba environments relatively small and atomic (e.g. an environment for each software package or for small sets of related packages).\nSingularity and Docker, on the other hand, allow for more complex environments. However, to create your own container requires more advanced knowledge and a steep learning curve. Fortunately, there are many existing containers available for bioinformatics, which Nextflow uses in its pipelines."
+ },
+ {
+ "objectID": "materials/05-software/01-managing_software.html#summary",
+ "href": "materials/05-software/01-managing_software.html#summary",
+ "title": "12 Managing Bioinformatics Software",
+ "section": "12.4 Summary",
+ "text": "12.4 Summary\n\n\n\n\n\n\nKey Points\n\n\n\n\nLinux is used in bioinformatics due to its open-source nature, powerful command-line interface, and compatibility with bioinformatics tools.\nPackage managers such as apt enable global software installations on Linux systems, simplifying the process of obtaining and managing software at the system level.\nLocal package managers such as mamba allow users to install and manage software within user-specific environments, avoiding conflicts with system-level packages.\nSoftware containers, such as Docker and Singularity, encapsulate entire environments in a file and can be used to manage more complex software enviroments.\nBioinformatics workflow managers such as Nextflow can make use of software containers to run complex bioinformatic pipelines."
+ },
+ {
+ "objectID": "materials/05-software/03-software_setup.html#sec-install-linux",
+ "href": "materials/05-software/03-software_setup.html#sec-install-linux",
+ "title": "13 Software setup",
+ "section": "13.1 Install Linux",
+ "text": "13.1 Install Linux\n\nFresh InstallationWindows WSLVirtual Machine\n\n\nThe recommendation for bioinformatic analysis is to have a dedicated computer running a Linux distribution. The kind of distribution you choose is not critical, but we recommend Ubuntu if you are unsure.\nYou can follow the installation tutorial on the Ubuntu webpage.\n\n\n\n\n\n\nWarning\n\n\n\nInstalling Ubuntu on the computer will remove any other operating system you had previously installed, and can lead to data loss.\n\n\n\n\nThe Windows Subsystem for Linux (WSL2) runs a compiled version of Ubuntu natively on Windows.\nThere are detailed instructions on how to install WSL on the Microsoft documentation page. But briefly:\n\nClick the Windows key and search for Windows PowerShell, right-click on the app and choose Run as administrator.\nAnswer “Yes” when it asks if you want the App to make changes on your computer.\nA terminal will open; run the command: wsl --install.\nProgress bars will show while installing “Virtual Machine Platform”, “Windows Subsystem for Linux” and finally “Ubuntu” (this process can take a long time).\n\nNote: it has happened to us in the past that the terminal freezes at the step of installing “Ubuntu”. If it is frozen for ~1h at that step, press Ctrl + C and hopefully you will get a message saying “Ubuntu installed successfully”.\n\nAfter installation completes, restart your computer.\nAfter restart, a terminal window will open asking you to create a username and password.\nIf it doesn’t, click the Windows key and search for Ubuntu, click on the App and it should open a new terminal.\n\nYou can use the same username and password that you have on Windows, or a different one - it’s your choice. Spaces and other special characters are not allowed for your Ubuntu username.\nNote: when you type your password nothing seems to be happening as the cursor doesn’t move. However, the terminal is recording your password as you type. You will be asked to type the new password again to confirm it, so you can always try again if you get it wrong the first time.\n\n\nYou should now have access to a Ubuntu Linux terminal. This behaves very much like a regular Ubuntu server.\n\nConfiguring WSL2\nAfter installation, it is useful to create shortcuts to your files on Windows. Your main C:\\ drive is located in /mnt/c/ and other drives will be equally available based on their letter. To create shortcuts to commonly-used directories you use symbolic links. Here are some commands to automatically create shortcuts to your Windows “Documents”, “Desktop” and “Downloads” folders (copy/paste these commands on the terminal):\nln -s $(wslpath $(powershell.exe '[environment]::getfolderpath(\"MyDocuments\")' | tr -d '\\r')) ~/Documents\nln -s $(wslpath $(powershell.exe '[environment]::getfolderpath(\"Desktop\")' | tr -d '\\r')) ~/Desktop\nln -s $(wslpath $(powershell.exe '[environment]::getfolderpath(\"UserProfile\")' | tr -d '\\r'))/Downloads ~/Downloads\nYou may also want to configure the Windows terminal to automatically open WSL2 (instead of the default Windows Command Prompt or Powershell):\n\nSearch for and open the “ Terminal” application.\nClick on the down arrow in the toolbar.\nClick on “ Settings”.\nUnder “Default Profile” select “ Ubuntu”.\n\n\n\n\nAnother way to run Linux within Windows (or macOS) is to install a Virtual Machine. However, this is mostly suitable for practicing and not suitable for real data analysis.\nDetails for installing Ubuntu on VirtualBox is given on this page. Make sure to do these things, while you are setting it up:\n\nIn Step 2 “Create a user profile”: make sure to tick the Guest Additions option.\nIn Step 2 “Define the Virtual Machine’s resources”:\n\nAssign at least 4 CPUs and 16000MB of RAM. At the very minimum you need 2 CPUs to run an Ubuntu VM.\nSet at least 100GB as disk size, more if you have it available (note, this will not take 100GB of space on your computer, but it will allow using up to a maximum of that value, which is useful as we are working with sequencing data).\n\n\nOnce the installation completes, login to the Ubuntu Virtual machine, open a terminal and do the following:\n\nRun su command.\nEnter your user password. Your terminal should change to start with root@\nType the command: usermod -a -G sudo YOUR-USERNAME-HERE.\nClose the terminal and restart the virtual machine.\n\nThese commands will add your newly created user to the “sudo” (admin) group.\n\n\n\nAfter making a fresh install of Ubuntu, open a terminal and run the following commands to update your system and install some essential packages:\nsudo apt update && sudo apt upgrade -y && sudo apt autoremove -y\nsudo apt install -y git\nsudo apt install -y default-jre"
+ },
+ {
+ "objectID": "materials/05-software/03-software_setup.html#conda",
+ "href": "materials/05-software/03-software_setup.html#conda",
+ "title": "13 Software setup",
+ "section": "13.2 Conda",
+ "text": "13.2 Conda\nWe recommend using the Conda package manager to install your software. In particular, the newest implementation called Mamba.\nTo install Mamba, run the following commands from the terminal:\nwget \"https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-$(uname)-$(uname -m).sh\"\nbash Miniforge3-$(uname)-$(uname -m).sh -b -p $HOME/miniforge3\nrm Miniforge3-$(uname)-$(uname -m).sh\n$HOME/miniforge3/bin/mamba init\nRestart your terminal (or open a new one) and confirm that your shell now starts with the word (base). Then run the following commands:\nRestart your terminal (or open a new one) and confirm that your shell now starts with the word (base). Then run the following commands:\nconda config --add channels defaults\nconda config --add channels bioconda\nconda config --add channels conda-forge\nconda config --set remote_read_timeout_secs 1000"
+ },
+ {
+ "objectID": "materials/05-software/03-software_setup.html#nextflow",
+ "href": "materials/05-software/03-software_setup.html#nextflow",
+ "title": "13 Software setup",
+ "section": "13.3 Nextflow",
+ "text": "13.3 Nextflow\nWe recommend that you install Nextflow within a conda environment. You can do that with the following command:\nmamba create -n nextflow -y nextflow\nWhen you want to use Nextflow make sure to activate this software environment by running mamba activate nextflow.\nAlso run the following command to create a configuration file to setup Nextflow correctly (make sure to copy all the code from top to bottom):\nmkdir -p $HOME/.nextflow\necho \"\nconda {\n conda.enabled = true\n singularity.enabled = false\n docker.enabled = false\n useMamba = true\n createTimeout = '4 h'\n cacheDir = '$HOME/.nextflow-conda-cache/'\n}\nsingularity {\n singularity.enabled = true\n conda.enabled = false\n docker.enabled = false\n pullTimeout = '4 h'\n cacheDir = '$HOME/.nextflow-singularity-cache/'\n}\ndocker {\n docker.enabled = true\n singularity.enabled = false\n conda.enabled = false\n}\n\" >> $HOME/.nextflow/config"
+ },
+ {
+ "objectID": "materials/05-software/03-software_setup.html#bioinformatics-software",
+ "href": "materials/05-software/03-software_setup.html#bioinformatics-software",
+ "title": "13 Software setup",
+ "section": "13.4 Bioinformatics Software",
+ "text": "13.4 Bioinformatics Software\nDue to conflicts in software versions required by different packages, we install them in separate environments:\nmamba create -n seqkit -y seqkit\nmamba create -n pangolin -y pangolin\nmamba create -n nextclade -y nextclade\nmamba create -n civet -y civet\nmamba create -n phylo -y mafft iqtree treetime figtree\nIn addition to these packages, you can also install AliView (which unfortunately is not available through Conda):\nwget https://ormbunkar.se/aliview/downloads/linux/linux-version-1.28/aliview.tgz\ntar -xzvf aliview.tgz\nrm aliview.tgz\necho \"alias aliview='java -jar $HOME/aliview/aliview.jar'\" >> $HOME/.bashrc\nFinally, you can install IGV by downloading the installer from their website."
+ },
+ {
+ "objectID": "materials/05-software/03-software_setup.html#software-image-containers",
+ "href": "materials/05-software/03-software_setup.html#software-image-containers",
+ "title": "13 Software setup",
+ "section": "13.5 Software Image Containers",
+ "text": "13.5 Software Image Containers\n\n13.5.1 Singularity\nWe recommend that you install Singularity and use the -profile singularity option when running Nextflow pipelines. On Ubuntu/WSL2, you can install Singularity using the following commands:\nsudo apt install -y runc cryptsetup-bin uidmap\nwget -O singularity.deb https://github.com/sylabs/singularity/releases/download/v4.0.2/singularity-ce_4.0.2-$(lsb_release -cs)_amd64.deb\nsudo dpkg -i singularity.deb\nrm singularity.deb\nIf you have a different Linux distribution, you can find more detailed instructions on the Singularity documentation page.\nIf you have issues running Nextflow pipelines with Singularity, then you can follow the instructions below for Docker instead.\n\n\n13.5.2 Docker\n\nWindows WSLLinux\n\n\nWhen using WSL2 on Windows, running Nextflow pipelines with -profile singularity sometimes doesn’t work.\nAs an alternative you can instead use Docker, which is another software containerisation solution. To set this up, you can follow the full instructions given on the Microsoft Documentation: Get started with Docker remote containers on WSL 2.\nWe briefly summarise the instructions here (but check that page for details and images):\n\nDownload Docker for Windows.\nRun the installer and install accepting default options.\nRestart the computer.\nOpen Docker and go to Settings > General to tick “Use the WSL 2 based engine”.\nGo to Settings > Resources > WSL Integration to enable your Ubuntu WSL installation.\n\nOnce you have Docker set and installed, you can use -profile docker when running your Nextflow command.\n\n\nFor Linux, here are the installation instructions:\nsudo apt install curl\ncurl -fsSL https://get.docker.com -o get-docker.sh\nsudo sh ./get-docker.sh\nsudo groupadd docker\nsudo usermod -aG docker $USER\nAfter the last step, you will need to restart your computer. From now on, you can use -profile docker when you run Nextflow"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#bam-binary-alignment-map",
+ "href": "materials/appendices/file_formats.html#bam-binary-alignment-map",
+ "title": "Common file formats",
+ "section": "BAM (“Binary Alignment Map”)",
+ "text": "BAM (“Binary Alignment Map”)\n\nBinary file.\nSame as a SAM file but compressed in binary form.\nFile extensions: .bam"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#bed-browser-extensible-data",
+ "href": "materials/appendices/file_formats.html#bed-browser-extensible-data",
+ "title": "Common file formats",
+ "section": "BED (“Browser Extensible Data”)",
+ "text": "BED (“Browser Extensible Data”)\n\nText file.\nStores coordinates of genomic regions.\nFile extension: .bed"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#csv-comma-separated-values",
+ "href": "materials/appendices/file_formats.html#csv-comma-separated-values",
+ "title": "Common file formats",
+ "section": "CSV (“Comma Separated Values”)",
+ "text": "CSV (“Comma Separated Values”)\n\nText file.\nStores tabular data in a text file. (also see TSV format)\nFile extensions: .csv\n\nThese files can be opened with spreadsheet programs (such as Microsoft Excel). They can also be created from spreadsheet programs by going to File > Save As… and select “CSV (Comma delimited)” as the file format."
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#fast5",
+ "href": "materials/appendices/file_formats.html#fast5",
+ "title": "Common file formats",
+ "section": "FAST5",
+ "text": "FAST5\n\nBinary file. More specifically, this is a Hierarchical Data Format (HDF5) file.\nUsed by Nanopore platforms to store the called sequences (in FASTQ format) as well as the raw electrical signal data from the pore.\nFile extensions: .fast5"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#fasta",
+ "href": "materials/appendices/file_formats.html#fasta",
+ "title": "Common file formats",
+ "section": "FASTA",
+ "text": "FASTA\n\nText file.\nStores nucleotide or amino acid sequences.\nFile extensions: .fa or .fas or .fasta"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#fastq",
+ "href": "materials/appendices/file_formats.html#fastq",
+ "title": "Common file formats",
+ "section": "FASTQ",
+ "text": "FASTQ\n\nText file, but often compressed with gzip.\nStores sequences and their quality scores.\nFile extensions: .fq or .fastq (compressed as .fq.gz or .fastq.gz)"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#gff-general-feature-format",
+ "href": "materials/appendices/file_formats.html#gff-general-feature-format",
+ "title": "Common file formats",
+ "section": "GFF (“General Feature Format”)",
+ "text": "GFF (“General Feature Format”)\n\nText file.\nStores gene coordinates and other features.\nFile extension: .gff"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#newick",
+ "href": "materials/appendices/file_formats.html#newick",
+ "title": "Common file formats",
+ "section": "NEWICK",
+ "text": "NEWICK\n\nText file.\nStores phylogenetic trees including nodes names and edge lengths.\nFile extensions: .tree or .treefile"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#sam-sequence-alignment-map",
+ "href": "materials/appendices/file_formats.html#sam-sequence-alignment-map",
+ "title": "Common file formats",
+ "section": "SAM (“Sequence Alignment Map”)",
+ "text": "SAM (“Sequence Alignment Map”)\n\nText file.\nStores sequences aligned to a reference genome. (also see BAM format)\nFile extensions: .sam"
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#tsv-tab-separated-values",
+ "href": "materials/appendices/file_formats.html#tsv-tab-separated-values",
+ "title": "Common file formats",
+ "section": "TSV (“Tab-Separated Values”)",
+ "text": "TSV (“Tab-Separated Values”)\n\nText file.\nStores tabular data in a text file. (also see CSV format)\nFile extensions: .tsv or .txt\n\nThese files can be opened with spreadsheet programs (such as Microsoft Excel). They can also be created from spreadsheet programs by going to File > Save As… and select “Text (Tab delimited)” as the file format."
+ },
+ {
+ "objectID": "materials/appendices/file_formats.html#vcf-variant-calling-format",
+ "href": "materials/appendices/file_formats.html#vcf-variant-calling-format",
+ "title": "Common file formats",
+ "section": "VCF (“Variant Calling Format”)",
+ "text": "VCF (“Variant Calling Format”)\n\nText file but often compressed with gzip.\nStores SNP/Indel variants\nFile extension: .vcf (or compressed as .vcf.gz)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#documentation-and-help",
+ "href": "materials/appendices/unix_cheatsheet.html#documentation-and-help",
+ "title": "Unix cheat sheet",
+ "section": "Documentation and Help",
+ "text": "Documentation and Help\n\n\n\n\n\n\n\nman {command}\nmanual page for the program\n\n\nwhatis {command}\nshort description of the program\n\n\n{command} --help\nmany programs use the --help flag to print documentation"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#listing-files",
+ "href": "materials/appendices/unix_cheatsheet.html#listing-files",
+ "title": "Unix cheat sheet",
+ "section": "Listing files",
+ "text": "Listing files\n\n\n\nls\nlist files in the current directory\n\n\nls {path}\nlist files in the specified path\n\n\nls -l {path}\nlist files in long format (more information)\n\n\nls -a {path}\nlist all files (including hidden files)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#change-directories",
+ "href": "materials/appendices/unix_cheatsheet.html#change-directories",
+ "title": "Unix cheat sheet",
+ "section": "Change Directories",
+ "text": "Change Directories\n\n\n\n\n\n\n\ncd {path}\nchange to the specified directory\n\n\ncd or cd ~\nchange to the home directory\n\n\ncd ..\nmove back one directory\n\n\npwd\nprint working directory. Shows the full path of where you are at the moment (useful if you are lost)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#make-or-remove-directories",
+ "href": "materials/appendices/unix_cheatsheet.html#make-or-remove-directories",
+ "title": "Unix cheat sheet",
+ "section": "Make or Remove Directories",
+ "text": "Make or Remove Directories\n\n\n\n\n\n\n\nmkdir {dirname}\ncreate a directory with specified name\n\n\nrmdir {dirname}\nremove a directory (only works if the directory is empty)\n\n\nrm -r {dirname}\nremove the directory and all it’s contents (use with care)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#copy-move-and-remove-files",
+ "href": "materials/appendices/unix_cheatsheet.html#copy-move-and-remove-files",
+ "title": "Unix cheat sheet",
+ "section": "Copy, Move and Remove Files",
+ "text": "Copy, Move and Remove Files\n\n\n\n\n\n\n\ncp {source/path/file1} {target/path/}\ncopy “file1” to another directory keeping its name\n\n\ncp {source/path/file1} {target/path/file2}\ncopy “file1” to another directory naming it “file2”\n\n\ncp {file1} {file2}\nmake a copy of “file1” in the same directory with a new name “file2”\n\n\nmv {source/path/file1} {target/path/}\nmove “file1” to another directory keeping its name\n\n\nmv {source/path/file1} {target/path/file2}\nmove “file1” to another directory renaming it as “file2”\n\n\nmv {file1} {file2}\nis equivalent to renaming a file\n\n\nrm {filename}\nremove a file"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#view-text-files",
+ "href": "materials/appendices/unix_cheatsheet.html#view-text-files",
+ "title": "Unix cheat sheet",
+ "section": "View Text Files",
+ "text": "View Text Files\n\n\n\n\n\n\n\nless {file}\nview and scroll through a text file\n\n\nhead {file}\nprint the first 10 lines of a file\n\n\nhead -n {N} {file}\nprint the first N lines of a file\n\n\ntail {file}\nprint the last 10 lines of a file\n\n\ntail -n {N} {file}\nprint the last N lines of a file\n\n\nhead -n {N} {file} | tail -n 1\nprint the Nth line of a file\n\n\ncat {file}\nprint the whole content of the file\n\n\ncat {file1} {file2} {...} {fileN}\nconcatenate files and print the result\n\n\nzcat {file} and zless {file}\nlike cat and less but for compressed files (.zip or .gz)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#find-patterns",
+ "href": "materials/appendices/unix_cheatsheet.html#find-patterns",
+ "title": "Unix cheat sheet",
+ "section": "Find Patterns",
+ "text": "Find Patterns\nFinding (and replacing) patterns in text is a very powerful feature of several command line programs. The patterns are specified using regular expressions (shortened as regex), which are not covered in this document. See this Regular Expressions Cheat Sheet for a comprehensive overview.\n\n\n\n\n\n\n\ngrep {regex} {file}\nprint the lines of the file that have a match with the regular expression pattern"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#wildcards",
+ "href": "materials/appendices/unix_cheatsheet.html#wildcards",
+ "title": "Unix cheat sheet",
+ "section": "Wildcards",
+ "text": "Wildcards\n\n\n\n\n\n\n\n*\nmatch any number of characters\n\n\n?\nmatch any character only once\n\n\nExamples\n\n\n\nls sample*\nlist all files that start with the word “sample”\n\n\nls *.txt\nlist all the files with .txt extension\n\n\ncp * {another/directory}\ncopy all the files in the current directory to a different directory"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#redirect-output",
+ "href": "materials/appendices/unix_cheatsheet.html#redirect-output",
+ "title": "Unix cheat sheet",
+ "section": "Redirect Output",
+ "text": "Redirect Output\n\n\n\n\n\n\n\n{command} > {file}\nredirect output to a file (overwrites if the file exists)\n\n\n{command} >> {file}\nappend output to a file (creates a new file if it does not already exist)"
+ },
+ {
+ "objectID": "materials/appendices/unix_cheatsheet.html#combining-commands-with-pipes",
+ "href": "materials/appendices/unix_cheatsheet.html#combining-commands-with-pipes",
+ "title": "Unix cheat sheet",
+ "section": "Combining Commands with | Pipes",
+ "text": "Combining Commands with | Pipes\n\n\n\n\n\n\n\n<command1> | <command2>\nthe output of “command1” is passed as input to “command2”\n\n\nExamples\n\n\n\nls | wc -l\ncount the number of files in a directory\n\n\ncat {file1} {file2} | less\nconcatenate files and view them with less\n\n\ncat {file} | grep \"{pattern}\" | wc -l\ncount how many lines in the file have a match with “pattern”"
+ },
+ {
+ "objectID": "materials/appendices/quick_r.html#rstudio",
+ "href": "materials/appendices/quick_r.html#rstudio",
+ "title": "R fundamentals",
+ "section": "RStudio",
+ "text": "RStudio\nR is the software and programming language itself, but the R interface is quite basic and not very user-friendly. Fortunately, there is a companion piece of software called RStudio, which makes working with R a little bit easier.\nThere are 4 basic panels in RStudio (see image below):\n\nThe script panel is essentially a text editor, where we can write code and save it as a text file, which in this case, because it contains R code, we call it an R script (but remember, a script is just a text file with some code in it. We’ve been creating shell scripts that run on the command line, here we have some R scripts, which contain R code)\nThe console panel is where the code actually runs, or is executed. This is equivalent to the terminal, on the command line. If we want to execute a line of code, then we need to run it on the console.\n\nOne nice feature of RStudio is that we can edit our code on the script panel and then run a line of code from the script on the console - it’s like copy/pasting that line of code from the script to the console. This makes working interactively with RStudio much easier, because we can edit our code in the script and run it as we go along.\n\nOn the top-right we have the Environment panel, which shows us objects that we create, that store information such as tables of data that we read into R.\nFinally, on the bottom-right there are a few tabs: a file browser (allowing us to see files in our computer), a plot display tab (for plots we generate) and a help tab to look at documentation.\n\n\n\nSetting RStudio\nBefore we start working with RStudio, it’s a good idea to change one of its default options. Go to Tools → Global Options… and change the following:\n\nThis will tell RStudio to NOT automatically save and load things that we have done in the past. You may think this is a helpful thing, but actually it’s very inconvenient, because if you are doing multiple analysis, it might get very very confusing what the objects that you created are! So, it’s always best to start R with a fresh session, and setting these options makes sure we do this.\n\n\nStarting a Project\nR has a concept called working directory, which is the location on your computer where it is working from (looking for files and folders). You can think of it as the folder that you cd into if you were working on the command-line.\nThe easiest way to ensure that R is using the correct working directory for our analysis, is to create an R project. In RStudio: File → New Project… → Existing Directory and then click the Browse… button to navigate to the folder where your project files are located.\nThis will create an .Rproj file on your project folder. Next time you want to work on your analysis, you can simply double-click on this file, and it will open RStudio, with the correct working directory already set for you."
+ },
+ {
+ "objectID": "materials/appendices/quick_r.html#r-basics",
+ "href": "materials/appendices/quick_r.html#r-basics",
+ "title": "R fundamentals",
+ "section": "R Basics",
+ "text": "R Basics\nThis section introduces some of the basic concepts in the R programming language.\n\nPackages/Libraries\nR has several extensions to the basic functionality called “packages” or “libraries”. A popular library for data manipulation is called tidyverse, which we are using in this course. Each time we start a new R session, we need to load the libraries we want to use:\n\nlibrary(tidyverse)\n\nIf you get an error like Error in library(tidyverse) : there is no package called 'tidyverse', that means that you didn’t install the package. To install packages you can run:\n\ninstall.packages(\"tidyverse\")\n\nYou only need to do this the first time you want to use a library. Once it’s installed, you don’t need to run this command again (unless you want to update the library to its latest version – often a good idea!).\n\n\nCreate objects\nCreate objects (something that contains a value) with <-. For example, the following creates an object called x containing a single number:\n\nx <- 53.341\n\nWe can print the content of the object by typing its name:\n\nx\n\n[1] 53.341\n\n\n\n\nFunctions\nMost of the tasks we can achieve in R are done through the use of functions. We can think of functions as mini-programs that take an input and give an output.\nFunctions are easy to identify, because they are always followed by parenthesis. Inside the parenthesis we include the inputs to the function.\n\nround(x) # round the the value of x\n\n[1] 53\n\n\nFunctions have options that can change their behaviour. Separate options using a comma:\n\nround(x, digits = 1) # round to one decimal point\n\n[1] 53.3\n\n\n\n\nVector\nA vector is the most basic type of object in R. It is a collection of values, which are all of the same type, for example numeric, character or logical (TRUE/FALSE).\n\nx_chr <- c(\"dog\", \"cat\", \"goldfish\") # character vector\nx_num <- c(1, 5, 23.3, 55.2) # numeric vector\nx_log <- c(TRUE, TRUE, FALSE, TRUE) # logical vector\n\nAccess values inside the vector with []:\n\nx_chr[2] # the second value\n\n[1] \"cat\"\n\nx_chr[c(2, 3)] # the second and third values\n\n[1] \"cat\" \"goldfish\"\n\n\n\n\nConditions\nIn many situations (for example to filter rows in a table), it’s useful to evaluate a set of conditions. We can create logical vectors using conditions:\n\nx_num\n\n[1] 1.0 5.0 23.3 55.2\n\n# is x_num greater than 20?\nx_num > 20\n\n[1] FALSE FALSE TRUE TRUE\n\n# is x_num equal to 5?\nx_num == 5\n\n[1] FALSE TRUE FALSE FALSE\n\n# is x_num contained the vector on the right?\nx_num %in% c(20, 30, 1)\n\n[1] TRUE FALSE FALSE FALSE\n\n\nCombine conditions with & (AND) and | (OR):\n\nx_num\n\n[1] 1.0 5.0 23.3 55.2\n\n# is x_num greater than or equal to 10 AND smaller than or equal to 30?\nx_num >= 10 & x_num <= 30\n\n[1] FALSE FALSE TRUE FALSE\n\n# is x_num smaller than 10 OR greater than 30?\nx_num < 10 | x_num > 30\n\n[1] TRUE TRUE FALSE TRUE\n\n\nTo set the filtering conditions, several relational operators can be used:\n\n== is equal to\n!= is different from\n%in% is contained in\n> is greater than\n>= is greater than or equal to\n< is less than\n<= is less than or equal to\n\nIt is also possible to combine several conditions together using the following logical operators:\n\n& AND\n| OR\n\n\n\nMissing Values\nSometimes we have missing values in our data, which are encoded as NA:\n\ny <- c(23, 44, NA, 212)\n\nWe need to ensure these are dealt with properly\n\n# returns NA\nmean(y)\n\n[1] NA\n\n# removes NA and then calculates mean\nmean(y, na.rm = TRUE)\n\n[1] 93\n\n\nThe is.na() function is important to deal with missing values:\n\ny\n\n[1] 23 44 NA 212\n\n# create a logical that is true if value is missing\nis.na(y)\n\n[1] FALSE FALSE TRUE FALSE\n\n# Negate that expression using !\n!is.na(y)\n\n[1] TRUE TRUE FALSE TRUE\n\n\n\n\nTables: data.frame/tibble\nTables in R are called data.frame. The tidyverse package has its own version of a data.frame called a tibble. For the most part they are basically equivalent, but the tibble object has a nicer printing function to display our data on the console.\nAs an example for working with tables in R, let’s read a TSV (tab-delimited) file that contains intervals of missing information in 5 SARS-CoV-2 consensus sequences (this data comes from the Switzerland case study). To import a TSV file into R as a data.frame we can use the function read_tsv() (for a CSV file we would use read_csv()):\n\nmissing_intervals <- read_tsv(\"missing_intervals.tsv\")\n\nRows: 132 Columns: 6\n── Column specification ────────────────────────────────────────────────────────\nDelimiter: \"\\t\"\nchr (4): seqID, patternName, pattern, strand\ndbl (2): start, end\n\nℹ Use `spec()` to retrieve the full column specification for this data.\nℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.\n\n\nWhen we read the table in, we get a message informing us of the column types found. In this case we have character columns containing text (indicated by chr) and numeric columns (indicated by dbl, which refers to the double-precission floating point format that computers use to store numbers).\nTo see the content of the table you can type the name of the object:\n\nmissing_intervals\n\n# A tibble: 132 × 6\n seqID patternName pattern strand start end\n <chr> <chr> <chr> <chr> <dbl> <dbl>\n 1 CH01 N+ N+ + 1 54\n 2 CH01 N+ N+ + 1193 1264\n 3 CH01 N+ N+ + 4143 4322\n 4 CH01 N+ N+ + 6248 6294\n 5 CH01 N+ N+ + 7561 7561\n 6 CH01 N+ N+ + 9243 9311\n 7 CH01 N+ N+ + 10367 10367\n 8 CH01 N+ N+ + 11361 11370\n 9 CH01 N+ N+ + 13599 13613\n10 CH01 N+ N+ + 16699 16758\n# ℹ 122 more rows\n\n\nSometimes a more convenient way is to click the name of the table on the environment, which opens a new tab to preview your data.\n\n\n\nData viewer in RStudio."
+ },
+ {
+ "objectID": "materials/appendices/quick_r.html#data-manipulation",
+ "href": "materials/appendices/quick_r.html#data-manipulation",
+ "title": "R fundamentals",
+ "section": "Data Manipulation",
+ "text": "Data Manipulation\nMost of the work you will do in R is with tables of data (data.frame/tibble objects). There are several ways to manipulate tables in R, but we will give a quick overview of the functionality available through the tidyverse collection of packages.\n\nBasic “verbs”\nThere’s a set of basic functions that can be thought of as “data manipulation verbs”. They are:\n\nmutate() → add a new column of modify an existing one.\nselect() → select columns from the table.\nfilter() → subset the rows from the table that fullfill a certain logical condition.\n\nHere are some examples of each:\n\n# create a new column with the missing interval lengths\nmutate(missing_intervals, \n length = (end - start) + 1)\n\n# A tibble: 132 × 7\n seqID patternName pattern strand start end length\n <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>\n 1 CH01 N+ N+ + 1 54 54\n 2 CH01 N+ N+ + 1193 1264 72\n 3 CH01 N+ N+ + 4143 4322 180\n 4 CH01 N+ N+ + 6248 6294 47\n 5 CH01 N+ N+ + 7561 7561 1\n 6 CH01 N+ N+ + 9243 9311 69\n 7 CH01 N+ N+ + 10367 10367 1\n 8 CH01 N+ N+ + 11361 11370 10\n 9 CH01 N+ N+ + 13599 13613 15\n10 CH01 N+ N+ + 16699 16758 60\n# ℹ 122 more rows\n\n# select only a few columns of the table\nselect(missing_intervals,\n seqID, start, end)\n\n# A tibble: 132 × 3\n seqID start end\n <chr> <dbl> <dbl>\n 1 CH01 1 54\n 2 CH01 1193 1264\n 3 CH01 4143 4322\n 4 CH01 6248 6294\n 5 CH01 7561 7561\n 6 CH01 9243 9311\n 7 CH01 10367 10367\n 8 CH01 11361 11370\n 9 CH01 13599 13613\n10 CH01 16699 16758\n# ℹ 122 more rows\n\n# subset the table to include only intervals within the Spike protein\nfilter(missing_intervals,\n start > 21563 & end < 25384)\n\n# A tibble: 15 × 6\n seqID patternName pattern strand start end\n <chr> <chr> <chr> <chr> <dbl> <dbl>\n 1 CH01 N+ N+ + 21621 21670\n 2 CH01 N+ N+ + 23480 23507\n 3 CH02 N+ N+ + 21620 21670\n 4 CH02 N+ N+ + 23480 23508\n 5 CH03 N+ N+ + 21621 21670\n 6 CH03 N+ N+ + 23480 23507\n 7 CH04 N+ N+ + 21620 21670\n 8 CH04 N+ N+ + 22240 22240\n 9 CH04 N+ N+ + 23480 23509\n10 CH04 N+ N+ + 24062 24395\n11 CH05 N+ N+ + 21620 22530\n12 CH05 N+ N+ + 23489 24709\n13 CH06 N+ N+ + 21605 22515\n14 CH06 N+ N+ + 23474 23525\n15 CH06 N+ N+ + 23721 24694\n\n\n\n\nPipes\nWe can chain multiple commands together using pipes (similarly to pipes in Unix). In R the pipe is represented by |> (or %>%). The way the pipe works is that the output of one function is sent as the input to the next function.\nTaking the examples from the previous section, we could chain all those commands like this:\n\nmissing_intervals |> \n mutate(length = (end - start) + 1) |> \n select(seqID, start, end, length)\n\n# A tibble: 132 × 4\n seqID start end length\n <chr> <dbl> <dbl> <dbl>\n 1 CH01 1 54 54\n 2 CH01 1193 1264 72\n 3 CH01 4143 4322 180\n 4 CH01 6248 6294 47\n 5 CH01 7561 7561 1\n 6 CH01 9243 9311 69\n 7 CH01 10367 10367 1\n 8 CH01 11361 11370 10\n 9 CH01 13599 13613 15\n10 CH01 16699 16758 60\n# ℹ 122 more rows\n\n\nIf you want to update the missing_intervals table, then you need to use <- at the beggining of the chain of pipes:\n\nmissing_intervals <- missing_intervals |> \n mutate(length = (end - start) + 1) |> \n select(seqID, start, end, length)\n\n\n\nGrouped Summaries\nWe can calculate summaries of the data (e.g. mean, standard deviation, maximum, minimum) per group (e.g. per sample) using a pair of functions together. For example:\n\n# mean and maximum interval length per sample\nmissing_intervals |> \n # for each sample\n group_by(seqID) |> \n # calculate summary statistics\n summarise(max_length = max(length),\n min_length = min(length),\n mean_length = mean(length))\n\n# A tibble: 7 × 4\n seqID max_length min_length mean_length\n <chr> <dbl> <dbl> <dbl>\n1 CH01 238 1 57 \n2 CH02 274 2 66 \n3 CH03 279 10 70.4\n4 CH04 1011 1 178. \n5 CH05 1221 11 200 \n6 CH06 974 1 166. \n7 CH07 5130 1 343. \n\n\nAs before, if we wanted to save this result in a new object, we would need to use <-:\n\nintervals_summary <- missing_intervals |> \n # for each sample\n group_by(seqID) |> \n # calculate summary statistics\n summarise(max_length = max(length),\n min_length = min(length),\n mean_length = mean(length)) |> \n # and rename the seqID column\n rename(sample = seqID)\n\nNotice in this case we also renamed the column called seqID to be named sample instead (this will be useful for the exercise later on).\nAnother useful function is count(), which counts how many times the values in a column appear on a data frame. For example, if we wanted to know how many missing intervals each sample had, we could do it like this:\n\nmissing_intervals |> \n count(seqID)\n\n# A tibble: 7 × 2\n seqID n\n <chr> <int>\n1 CH01 16\n2 CH02 19\n3 CH03 16\n4 CH04 21\n5 CH05 15\n6 CH06 16\n7 CH07 29\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nModify the following code:\n\nmissing_intervals |> \n count(seqID)\n\nTo also:\n\nrename the column seqID to be named sample instead.\nsave the output in an object called intervals_count.\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nTo rename the column, we could use the rename() function:\n\nmissing_intervals |> \n count(seqID) |> \n rename(sample = seqID)\n\n# A tibble: 7 × 2\n sample n\n <chr> <int>\n1 CH01 16\n2 CH02 19\n3 CH03 16\n4 CH04 21\n5 CH05 15\n6 CH06 16\n7 CH07 29\n\n\nAnd to save this output to a new object, we need to use <-:\n\nintervals_count <- missing_intervals |> \n count(seqID) |> \n rename(sample = seqID)\n\n\n\n\n\n\n\n\n\n\n\n\n\nJoining Tables\nWe can join multiple tables together based on a common identifier. There are different types of join operations, depending on what we want to achieve.\nTake these two tables as an example (these tables come pre-loaded with tidyverse):\n\nband_members\n\n# A tibble: 3 × 2\n name band \n <chr> <chr> \n1 Mick Stones \n2 John Beatles\n3 Paul Beatles\n\nband_instruments\n\n# A tibble: 3 × 2\n name plays \n <chr> <chr> \n1 John guitar\n2 Paul bass \n3 Keith guitar\n\n\nHere are some different ways we can join these tables:\n\n# keep all records from both tables\nfull_join(band_members, band_instruments, by = \"name\")\n\n# A tibble: 4 × 3\n name band plays \n <chr> <chr> <chr> \n1 Mick Stones <NA> \n2 John Beatles guitar\n3 Paul Beatles bass \n4 Keith <NA> guitar\n\n# keep all records from the first table\nleft_join(band_members, band_instruments, by = \"name\")\n\n# A tibble: 3 × 3\n name band plays \n <chr> <chr> <chr> \n1 Mick Stones <NA> \n2 John Beatles guitar\n3 Paul Beatles bass \n\n# keep all records from the second table\nright_join(band_members, band_instruments, by = \"name\")\n\n# A tibble: 3 × 3\n name band plays \n <chr> <chr> <chr> \n1 John Beatles guitar\n2 Paul Beatles bass \n3 Keith <NA> guitar\n\n# keep only the records occurring in both tables\ninner_join(band_members, band_instruments, by = \"name\")\n\n# A tibble: 2 × 3\n name band plays \n <chr> <chr> <chr> \n1 John Beatles guitar\n2 Paul Beatles bass \n\n\nIn each case, if there was no match between the two tables, the cells are filled with missing values NA.\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nIn this exercise we will join the intervals_summary table we created earlier, with the metadata table that contains information about our samples.\n\nRead the your metadata table sample_info.csv into R and save it as an object called sample_info.\n\n\nHint\n\nRemember that you can use the read_csv() function to read CSV files into R.\n\nJoin the sample_info table that you just imported with the intervals_summary table we created earlier. Save the output to the intervals_summary table (this will update the table).\n\n\nYou can use the left_join() function to achieve this, using the “sample” column as the identifier column used to join the two tables.\n\n\n\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nAnswer 1\nTo read the metadata CSV file, we use the read_csv() function, and use <- to save the output in an object called sample_info:\n\nsample_info <- read_csv(\"sample_info.csv\")\n\nRows: 7 Columns: 4\n── Column specification ────────────────────────────────────────────────────────\nDelimiter: \",\"\nchr (3): sample, country, sequencing_instrument\ndate (1): collection_date\n\nℹ Use `spec()` to retrieve the full column specification for this data.\nℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.\n\n\nWe can see the content of our file by typing its name in the console. Here is the file in our example data (yours might look different):\n\nsample_info\n\n# A tibble: 7 × 4\n sample collection_date country sequencing_instrument\n <chr> <date> <chr> <chr> \n1 CH01 2021-11-09 Switzerland GridION \n2 CH04 2021-12-20 Switzerland GridION \n3 CH05 2021-12-20 Switzerland GridION \n4 CH06 2021-12-20 Switzerland GridION \n5 CH07 2021-12-20 Switzerland GridION \n6 CH02 2021-12-21 Switzerland GridION \n7 CH03 2021-12-22 Switzerland GridION \n\n\nAnswer 2\nTo join the two tables together, we can use one of the *_join() functions. In this case it doesn’t matter which function we use, because both tables have the same sample IDs. But, for example, let’s say we only wanted to retain the samples that are in common across both tables. In that case, we would use inner_join():\n\nintervals_summary <- inner_join(intervals_summary, sample_info, by = \"sample\")"
+ },
+ {
+ "objectID": "materials/appendices/quick_r.html#data-visualisation",
+ "href": "materials/appendices/quick_r.html#data-visualisation",
+ "title": "R fundamentals",
+ "section": "Data Visualisation",
+ "text": "Data Visualisation\nFor this section, we will use another table, which contains some of the metrics that we can collect from our consensus pipeline:\n\n# read the table\nqc_metrics <- read_tsv(\"consensus_metrics.tsv\")\n\nRows: 7 Columns: 6\n── Column specification ────────────────────────────────────────────────────────\nDelimiter: \"\\t\"\nchr (3): sample, qc_status, lineage\ndbl (3): n_mapped_reads, median_depth, pct_missing\n\nℹ Use `spec()` to retrieve the full column specification for this data.\nℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.\n\n# preview the table\nqc_metrics\n\n# A tibble: 7 × 6\n sample n_mapped_reads median_depth pct_missing qc_status lineage\n <chr> <dbl> <dbl> <dbl> <chr> <chr> \n1 CH01 43900 248 3.05 good BA.1 \n2 CH02 42841 159 4.20 good BA.1 \n3 CH03 40079 160 3.77 good BA.1 \n4 CH04 50902 164 12.5 bad BA.1 \n5 CH05 32020 133 10.0 bad BA.1 \n6 CH06 46277 177 8.87 mediocre BA.1.1 \n7 CH07 15867 41 33.2 bad None \n\n\nWe can build plots from our tables using the ggplot2 package (which is also part of the tidyverse).\nTo build a ggplot, we usually need at least three things:\n\nThe data frame we want to use for the plot (the data).\nThe columns we want to visualise as our x-axis, y-axis, and colours (these are called aesthetics).\nThe type of shape that we want to plot (these are called geometries).\n\nFor example, let’s try to make a plot to show the relationship between total number of counts and the median depth of sequencing in these samples:\n\nggplot(data = qc_metrics, aes(x = n_mapped_reads, y = median_depth))\n\n\n\n\nWhen we do this, we simply get an empty plot, with x and y axis, but nothing drawn on it. To draw something on the plot, we add (literally with +) geometries to our plot. In this case, we can use the geom_point() geometry, which draws “points” on the plot:\n\nggplot(data = qc_metrics, aes(x = n_mapped_reads, y = median_depth)) +\n geom_point()\n\n\n\n\nThere are many geometries available with ggplot:\n\ngeom_point() draws points.\ngeom_boxplot() draws a boxplot.\ngeom_histogram() draws a histogram.\ngeom_col() draws a barplot (this one is named a little strangely, but “col” means it draws “columns” or bars).\n\nWe can further modify the look to the plot by adding other aesthetics such as the colour of the points. For example, let’s say we wanted to colour our points according to their “QC Status”:\n\nggplot(data = qc_metrics, \n aes(x = n_mapped_reads, y = median_depth, colour = qc_status)) +\n geom_point()\n\n\n\n\nFinally, we may sometimes want to change the labels of our plot. In that case, we can add the labs() function to our plotting code:\n\nggplot(data = qc_metrics, \n aes(x = n_mapped_reads, y = median_depth, colour = qc_status)) +\n geom_point() +\n labs(x = \"Number Mapped Reads\", \n y = \"Median Sequencing Depth\", \n colour = \"Nextclade QC Status\")\n\n\n\n\n\n\n\n\n\n\nExercise\n\n\n\n\n\n\n\nModify the plot we just did, to show the relationship between median depth of sequencing (x-axis) and percentage of missing bases (y-axis). Colour the points according to the lineage column.\n\n\n\n\n\n\nAnswer\n\n\n\n\n\n\n\nHere is the modified code:\n\nggplot(data = qc_metrics, \n aes(x = median_depth, y = pct_missing, colour = lineage)) +\n geom_point() +\n labs(x = \"Median Sequencing Depth\", \n y = \"% Missing Bases\", \n colour = \"Pango Lineage\")\n\n\n\n\nThe things we have changed were the aesthetics and labels."
+ },
+ {
+ "objectID": "materials/appendices/tools_and_resources.html#amplicon-primer-schemes",
+ "href": "materials/appendices/tools_and_resources.html#amplicon-primer-schemes",
+ "title": "Tools and Resources",
+ "section": "Amplicon Primer Schemes",
+ "text": "Amplicon Primer Schemes\nFor amplicon sequencing, there are several protocols and commercial kits available. We try to summarise some of the common ones below.\n\nARTIC\n\nV5.3.2V4.1V4V3V2V1Midnight\n\n\nARTIC primer scheme version 5.3.2 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 5.3.2 \\\n--schema_ignore_params 'genomes,primer_set_version'\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V5.3.2/SARS-CoV-2.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V5.3.2/SARS-CoV-2.scheme.bed'\n\n\nARTIC primer scheme version 4.1 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 4.1\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V4.1/SARS-CoV-2.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V4.1/SARS-CoV-2.scheme.bed'\n\n\nARTIC primer scheme version 4 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 4\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V4/SARS-CoV-2.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V4/SARS-CoV-2.scheme.bed'\n\n\nARTIC primer scheme version 3 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 3\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V3/nCoV-2019.primer.bed'\n\n\nARTIC primer scheme version 2 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 2\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V2/nCoV-2019.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V2/nCoV-2019.primer.bed'\n\n\nARTIC primer scheme version 1 (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 1\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V1/nCoV-2019.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/artic-network/artic-ncov2019/raw/master/primer_schemes/nCoV-2019/V1/nCoV-2019.primer.bed'\n\n\nPrimers for the “Midnight” protocol, also known as “1200” as it produces fragments that are ~1200bp long. This primer scheme is optimised for ONT platforms (link).\nTo analyse with nf-core/viralrecon, add these options to the command:\n--genome 'MN908947.3' \\\n--primer_set artic \\\n--primer_set_version 1200\nOr, alternatively, you can use the direct links to the FASTA/BED files:\n--fasta 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/primer_schemes/artic/nCoV-2019/V1200/nCoV-2019.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/primer_schemes/artic/nCoV-2019/V1200/nCoV-2019.bed'\n\n\n\n\n\nNEB VarSkip\n\n1a2a2bLong 1a\n\n\nNEB’s VarSkip kit, primer version 1a (link).\nFor analysis with nf-core/viralrecon the direct link to the FASTA and BED files can be given:\n--fasta 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V1a/NEB_VarSkip.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V1a/NEB_VarSkip.scheme.bed'\n\n\nNEB’s VarSkip kit, primer version 2a (link).\nFor analysis with nf-core/viralrecon the direct link to the FASTA and BED files can be given:\n--fasta 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V2a/NEB_VarSkip.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V2a/NEB_VarSkip.scheme.bed'\n\n\nNEB’s VarSkip kit, primer version 2b (link).\nFor analysis with nf-core/viralrecon the direct link to the FASTA and BED files can be given:\n--fasta 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V2b/NEB_VarSkip.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V2b/NEB_VarSkip.scheme.bed'\n\n\nNEB’s VarSkip kit, primer version 1a, long primers (link).\nFor analysis with nf-core/viralrecon the direct link to the FASTA and BED files can be given:\n--fasta 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V1a-long/NEB_VarSkip.reference.fasta' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/MN908947.3/GCA_009858895.3_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://raw.githubusercontent.com/nebiolabs/VarSkip/main/schemes/NEB_VarSkip/V1a-long/NEB_VarSkip.scheme.bed'\n\n\n\n\n\nAtoplex\nATOPlex kit.\nFor analysis with nf-core/viralrecon the direct link to the FASTA and BED files can be given:\n--fasta 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/NC_045512.2/GCF_009858895.2_ASM985889v3_genomic.200409.fna.gz' \\\n--gff 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/NC_045512.2/GCF_009858895.2_ASM985889v3_genomic.200409.gff.gz' \\\n--primer_bed 'https://github.com/nf-core/test-datasets/raw/viralrecon/genome/NC_045512.2/amplicon/nCoV-2019.atoplex.V1.bed'\n\n\nIllumina COVIDseq\n\nCOVIDseqCOVIDseq (RUO version)\n\n\nThe original COVIDseq kit uses ARTIC V3 primers.\nSee section above for nf-core/viralrecon options.\n\n\nThe COVIDseq RUO version uses ARTIC V4 primers.\nSee section above for nf-core/viralrecon options.\n\n\n\n\n\nSWIFT / xGEN\nThe “xGen SARS-CoV-2 Amplicon Panels”, previously called “SWIFT Normalase”, is commercialised by IDT. Unfortunately IDT does not make their BED files available publicly. If you purchase a kit from IDT, the company should provide you with the correct BED file to process your samples.\nNOTE: in the future we will provide a link to a BED file with approximate coordinates, inferred bioinformatically. This can be useful if re-analysing public data."
+ },
+ {
+ "objectID": "materials/appendices/tools_and_resources.html#data-resources",
+ "href": "materials/appendices/tools_and_resources.html#data-resources",
+ "title": "Tools and Resources",
+ "section": "Data Resources",
+ "text": "Data Resources\n\noutbreak.info - interactive exploration of global SARS-CoV-2 data.\nNextstrain SARS-CoV-2 resources - analysis and datasets curated by the Nextrain team.\nData portals:\n\nThe European COVID-19 Data Platform\nNCBI SARS-CoV-2 Resources\nGISAID\n\nUK-specific resources:\n\nCOVID-19 Genomics UK Consortium\nCOVID–19 Genomic Surveillance built by the Wellcome Sanger Institute."
+ },
+ {
+ "objectID": "materials/appendices/tools_and_resources.html#web-tools",
+ "href": "materials/appendices/tools_and_resources.html#web-tools",
+ "title": "Tools and Resources",
+ "section": "Web Tools",
+ "text": "Web Tools\n\nTaxonium - a tool for large tree visualisation."
+ },
+ {
+ "objectID": "materials/appendices/tools_and_resources.html#other-courses",
+ "href": "materials/appendices/tools_and_resources.html#other-courses",
+ "title": "Tools and Resources",
+ "section": "Other Courses",
+ "text": "Other Courses\n\nMutation calling, viral genome reconstruction and lineage/clade assignment from SARS-CoV-2 sequencing data - using the Galaxy platform.\nThe Power of Genomics to Understand the COVID-19 Pandemic - a general introduction to the COVID-19 pandemic.\nCOVID-10 Data Analysis - ARTIC and CLIMB-BIG-DATA joint workshop with a series of lectures and tutorials available."
+ }
+]
\ No newline at end of file
diff --git a/setup.html b/setup.html
new file mode 100644
index 0000000..e1dc8aa
--- /dev/null
+++ b/setup.html
@@ -0,0 +1,812 @@
+
+
+
+
+
+
+
+
+
+SARS Genomic Surveillance
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Data & Software
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+Workshop Attendees
+
+
+
+
If you are attending one of our workshops, we will provide a training environment with all of the required software and data. You do not need to download or install anything ahead of the workshop.
+If you want to setup your own computer to run the analysis demonstrated on this course, you can follow the instructions below.
+
+
+
+
Software
+
The software installation for this course is quite complex and detailed in a separate section.
+
+
+
Data
+
Different datasets are used throughout these materials. This page provides links to download each dataset, with a brief description for each of them. We have split the data between sequencing platforms.
These samples were downloaded from SRA and include samples collected between Nov and Dec 2021 in South Africa. These data are used in the worked example South Africa (Illumina).
+
We provide two versions of the data: the full data includes 24 samples (more realistic sample size), whereas the small version includes only 8 samples (quicker processing time for practising).
The standard panels for External Quality Assessment provided by NEQAS were sequenced on an Illumina platform by UKHSA and used to generate four sets of data for training purposes. These data can be used for the extended exercise: EQA (Exercise).
+
We provide a link to each of the four datasets below (the datasets are very similar to each other):
Nanopore chemistry: not specified (we assumed 9.4.1)
+
Primer set: not specified (we assumed ARTIC primers v3)
+
Guppy version: not specified (we assumed 3.6 or higher)
+
Basecalling mode: not specified (we assumed “high”)
+
+
+
Note that several details needed to run the medaka pipeline were not provided in the public repository. We arbitrarily picked parameters as detailed above, but this is for demonstration purposes only.
+
+
+Click here to see details of how the data was downloaded from public repositories
+
+
We obtained these data from the SRA repository, using the fastq-dump command as follows:
These samples were downloaded from SRA and include samples collected between Nov 2021 and Jan 2022 in Switzerland and sequenced by the Institute for Infectious Diseases, University of Bern. These data are used in the worked example Switzerland (Nanopore).
+
We provide two versions of the data: the full data includes 65 samples (more realistic sample size), whereas the small version includes only 10 samples (quicker processing time for practising).
Nanopore chemistry: not specified (we assumed 9.4.1)
+
Primer set: ARTIC primers v3
+
Guppy version: not specified (we assumed 3.6 or higher)
+
Basecalling mode: not specified (we assumed “fast”)
+
+
+
Note that several details needed to run the medaka pipeline were not provided in the public repository. We arbitrarily picked parameters as detailed above, but this is for demonstration purposes only.
+
+
+Click here to see details of how the data was downloaded from public repositories
+
+
We obtained these data from the SRA repository, using the fastq-dump command as follows:
The standard panels for External Quality Assessment provided by NEQAS were sequenced on an ONT platform by TODO. These data can be used for the extended exercise: EQA (Exercise).
+
We provide a link to datasets corresponding to two different runs: